
| 📌 The essentials On May 14, 2026, the FDA approved Trimbow (beclomethasone dipropionate/formoterol fumarate/glycopyrrolate, Chiesi USA), a single-inhaler triple combination therapy for the long-term maintenance treatment of asthma in adults. It is the first and currently only inhaled triple combination therapy approved in the United States for asthma that delivers all three drug classes, an inhaled corticosteroid (ICS), a long-acting beta2-agonist (LABA), and a long-acting muscarinic antagonist (LAMA), in a single pressurized metered-dose inhaler. Indication: long-term maintenance treatment of asthma in adults. Not for children. Not for use as a rescue inhaler. Dosing: 2 inhalations twice daily (morning and evening), 4 total per day. Two strengths: 86 mcg/4.9 mcg/10.6 mcg and 172 mcg/4.9 mcg/10.6 mcg per actuation. The clinical basis: two Phase 3 randomized, double-blind, active-controlled trials, TRIMARAN and TRIGGER, enrolling 2,592 adults with uncontrolled asthma. Both trials showed statistically significant improvements in pre-dose FEV1 at 26 weeks compared with dual ICS/LABA therapy alone. TRIMARAN also showed a statistically significant 15% reduction in the rate of moderate and severe exacerbations. Critical safety note: Trimbow contains a LABA (formoterol). Do not use any other LABA or any other anticholinergic medication while taking Trimbow. |
|---|
Asthma affects approximately 27 million Americans and is a leading cause of emergency department visits, missed school and work days, and chronic respiratory morbidity. For most patients, the treatment goal is straightforward: control inflammation with an inhaled corticosteroid, open the airways with a bronchodilator, and live without symptoms. For many, that goal is achievable with a combination ICS/LABA inhaler.
But a substantial proportion of patients, estimated at 5 to 10% of all asthma patients and sometimes characterized as moderate to severe uncontrolled asthma, do not achieve adequate control on dual ICS/LABA therapy even with appropriate adherence and technique. For these patients, the next therapeutic step has typically involved adding a long-acting muscarinic antagonist (LAMA), a third drug class that relaxes airway smooth muscle through a different mechanism than beta-agonists. Until now, adding a LAMA meant adding a separate inhaler, which adds complexity, cost, and another opportunity for missed or incorrect doses.
On May 14, 2026, the FDA approved Trimbow, a single inhaler containing all three classes simultaneously. The clinical data supporting it has been in the literature since 2019, published in The Lancet. The U.S. approval now makes this approach officially available to American patients and prescribers.
What Uncontrolled Asthma on ICS/LABA Looks Like and Why a Third Drug Class Helps
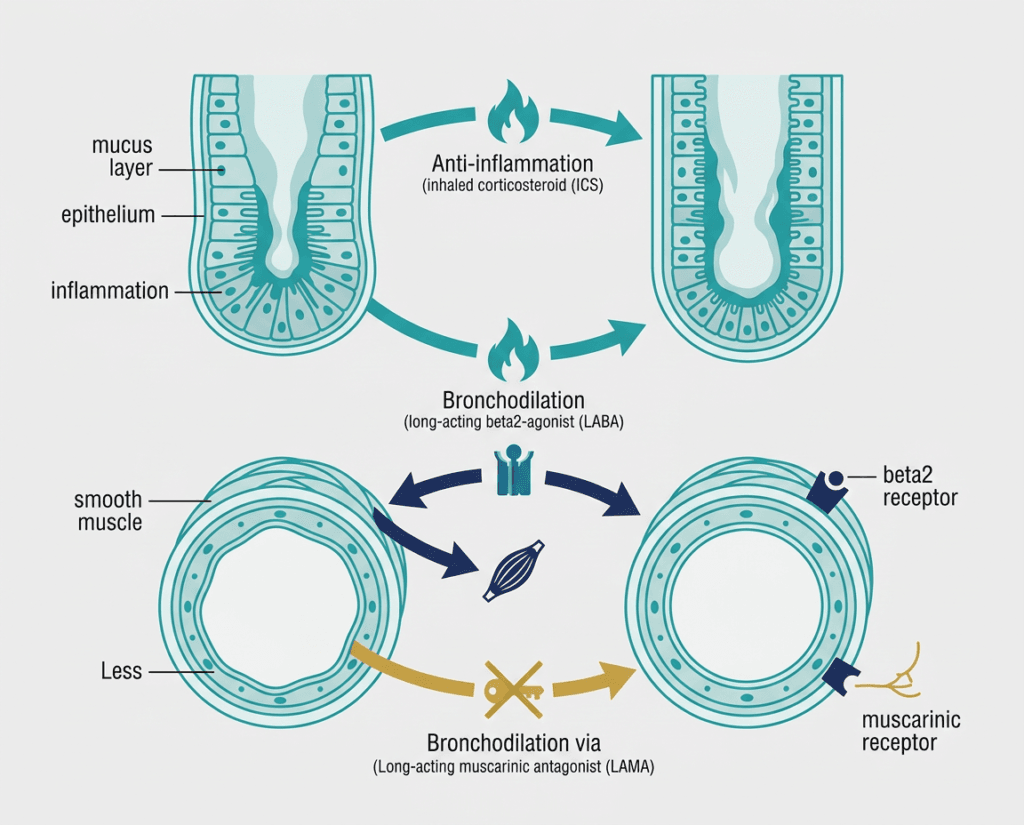
To understand what Trimbow adds, it helps to understand the three drug classes it combines and why each contributes something the others cannot fully replace.
Beclomethasone dipropionate: the ICS component
Beclomethasone dipropionate (BDP) is a synthetic glucocorticoid that suppresses the airway inflammation that underlies asthma. Inflammation is the root cause of the chronic bronchial hyperreactivity that makes asthmatic airways prone to spasm, swelling, and mucus production. ICS therapy is the foundational treatment for persistent asthma because no bronchodilator, however effective at opening airways acutely, addresses the underlying inflammatory state. BDP is one of the most extensively studied and commonly used ICS agents. In Trimbow, it is formulated in an extrafine particle size (mass median aerodynamic diameter below 2 micrometers) that allows improved deposition in the small airways, a region of the lung that is a significant site of inflammation in asthma and that larger-particle formulations reach less effectively.
Formoterol fumarate: the LABA component
Formoterol fumarate is a long-acting beta2-agonist (LABA) that works by binding to beta2-adrenergic receptors on airway smooth muscle, causing relaxation and dilation. This bronchodilation reduces airway resistance and improves airflow. Unlike short-acting beta2-agonists (SABAs) such as albuterol, which last 4 to 6 hours and are used for quick relief, formoterol is designed for sustained bronchodilation over approximately 12 hours. It is taken twice daily as a maintenance medication, not for acute symptoms. Formoterol also has a relatively rapid onset compared to other LABAs, beginning to act within minutes of inhalation.
An important safety note: LABAs, when used without an ICS, increase the risk of asthma-related hospitalizations and death. This risk drove FDA requirements that LABAs only be prescribed in combination with ICS for asthma. Trimbow contains both, so this risk is not present in the way it is with LABA-only products.
Glycopyrrolate: the LAMA component
Glycopyrrolate (also called glycopyrronium) is a long-acting muscarinic antagonist (LAMA) that blocks muscarinic receptors on airway smooth muscle and submucosal glands. Muscarinic receptors respond to acetylcholine, a neurotransmitter released by the parasympathetic nervous system. Parasympathetic stimulation causes bronchoconstriction and increased mucus secretion, both of which worsen airway obstruction in asthma. Glycopyrrolate blocks these effects, producing additional bronchodilation and reducing mucus production through a mechanism completely distinct from formoterol’s beta-agonist pathway.
This mechanistic complementarity is the rationale for combining a LABA and a LAMA: they dilate airways through two separate receptor pathways, producing additive bronchodilation beyond what either achieves alone. In COPD, where LABA/LAMA combinations are well-established, this additive effect is substantial. In asthma, the role of LAMA therapy is newer and the evidence base is still evolving, which is what makes the TRIMARAN and TRIGGER trial data important.
| Why adding a LAMA to asthma therapy is different from COPD In COPD, LAMA therapy is foundational and often used as first-line bronchodilation, because the disease is primarily driven by fixed airflow limitation and parasympathetically mediated bronchoconstriction. In asthma, the primary problem is ICS-reversible inflammation with episodic bronchospasm, and the role of the parasympathetic nervous system is less dominant than in COPD. This is why LAMAs were not part of asthma therapy for many years: the theoretical rationale was weaker. The TRIMARAN and TRIGGER trials were specifically designed to test whether adding a LAMA to ICS/LABA in patients already failing dual therapy would produce meaningful additional benefit. The answer, at least for lung function and for severe exacerbations in TRIMARAN, was yes. The question of which patients benefit most, and how much, is one the subgroup analyses have tried to answer. |
|---|
The TRIMARAN and TRIGGER Trials: What the Evidence Actually Shows
The FDA approval is based on two Phase 3 randomized, double-blind, active-controlled trials, both published in The Lancet in November 2019 and extensively analyzed in subsequent publications.
Trial design
Both trials enrolled adults with uncontrolled asthma, defined as:
- Asthma Control Questionnaire-7 (ACQ-7) score of 1.5 or higher (indicating inadequate asthma control)
- Pre-bronchodilator FEV1 below 80% of predicted normal (confirming measurable airflow limitation)
- History of at least one moderate or severe asthma exacerbation in the prior year (establishing meaningful disease burden)
- Current treatment with medium-dose ICS/LABA (TRIMARAN) or high-dose ICS/LABA (TRIGGER)
TRIMARAN NCT02676076: enrolled 1,155 patients. Compared medium-strength BDP/FF/G (100 mcg/6 mcg/10 mcg per actuation) versus medium-strength BDP/FF (100 mcg/6 mcg) for 52 weeks.
TRIGGER NCT02676089: enrolled 1,437 patients. Compared high-strength BDP/FF/G (200 mcg/6 mcg/10 mcg) versus high-strength BDP/FF (200 mcg/6 mcg), with an additional open-label arm receiving high-strength BDP/FF plus tiotropium (a separate LAMA) once daily, providing a comparison against adding a LAMA in a separate inhaler rather than a combined one.
Co-primary endpoints for both trials were pre-dose FEV1 at week 26 and the rate of moderate and severe exacerbations over 52 weeks.
Primary endpoint results
| Outcome | TRIMARAN (medium dose) | TRIGGER (high dose) |
|---|---|---|
| Improvement in pre-dose FEV1 at Week 26 (BDP/FF/G vs BDP/FF) | +57 mL (95% CI 15 to 99; p=0.0080) | +73 mL (95% CI 26 to 120; p=0.0025) |
| Rate of moderate-to-severe exacerbations over 52 weeks (rate ratio vs BDP/FF) | Rate ratio 0.85 (95% CI 0.73 to 0.99; p=0.033), 15% reduction | Rate ratio 0.88 (95% CI 0.75 to 1.03; p=0.11), 12% reduction, not statistically significant |
| Patients experiencing moderate-to-severe exacerbations | 58.6% (BDP/FF/G) vs 66.0% (BDP/FF) | 56.6% (BDP/FF/G) vs 63.7% (BDP/FF) |
| Patients experiencing severe exacerbations | 18.2% (BDP/FF/G) vs 22.4% (BDP/FF) | Numerically reduced; statistically significant for severe exacerbations |
| Tiotropium add-on vs BDP/FF/G (TRIGGER only) | — | No statistically significant differences between the combined inhaler and the separate LAMA arm |
The FEV1 improvements in both trials are statistically significant and, while modest in absolute terms (57 to 73 mL), are consistent with meaningful clinical benefit in a population where gains are incremental. The 15% reduction in moderate and severe exacerbation rate in TRIMARAN is the more clinically impactful finding: exacerbations are the main driver of asthma-related hospitalizations, emergency visits, oral corticosteroid courses, and lung function decline over time. Reducing exacerbations reduces downstream harm.
The fact that TRIGGER’s exacerbation reduction did not reach statistical significance (p=0.11 for combined moderate and severe) is worth acknowledging honestly. However, the point estimate was similar to TRIMARAN (12% vs 15%), and the severe exacerbation data in TRIGGER was statistically significant. The likely explanation for the differing statistical outcomes is that TRIGGER enrolled patients on higher-dose ICS/LABA, who are by definition more disease-refractory, making further improvement harder to demonstrate and more heterogeneous.
The TRIGGER tiotropium add-on arm is also clinically important. The three-way comparison showed no statistically significant difference in outcomes between the triple combination inhaler and adding tiotropium as a separate inhaler to ICS/LABA. This means Trimbow is not demonstrably superior to the established approach of adding a separate LAMA inhaler. What it offers is the same clinical effect in a single device rather than two, which is a convenience and adherence advantage rather than a pharmacological superiority.
Remission-on-treatment analysis
A post-hoc analysis published in the Journal of Allergy and Clinical Immunology (2025) evaluated whether triple therapy increased the proportion of patients achieving on-treatment remission, defined as no severe exacerbations and no systemic corticosteroid use over 52 weeks, plus ACQ-5 score below 1.5 at weeks 26, 40, and 52, plus stable or improved lung function. In TRIMARAN, 30.2% of patients on triple therapy met all remission criteria compared with 25.6% on dual therapy. This finding is exploratory and post-hoc, but it provides useful context for the meaningful goal of deep disease control rather than simply symptom reduction.
The Extrafine Formulation: Why Particle Size Matters
Trimbow is specifically described as an extrafine formulation, meaning the active ingredients are delivered in particles with a mass median aerodynamic diameter below 2 micrometers. This is smaller than standard inhaled corticosteroid particles, which typically have diameters of 2 to 5 micrometers.
The significance of small-particle size relates to where in the lung the drug deposits. Larger particles tend to deposit in the central airways, the larger bronchi visible on bronchoscopy. Smaller particles travel further into the lung, reaching the small airways below 2 mm in diameter. In asthma, inflammation and airflow limitation are present throughout the bronchial tree, including in these small peripheral airways, and conventional larger-particle inhalers may not adequately treat this compartment.
This is not a theoretical claim for Trimbow specifically: the TRIMARAN and TRIGGER trials were conducted with the extrafine formulation, and the results reflect its performance. Whether the extrafine delivery is responsible for a meaningful component of the observed benefit, or whether standard-particle LABA/LAMA/ICS would perform similarly, has not been tested head-to-head in asthma.
Who Is Trimbow For? Patient Selection and the Treatment Hierarchy
Trimbow is not a first-line therapy for asthma. It is indicated for adults with asthma that is not adequately controlled on existing treatment, specifically on medium or high-dose ICS/LABA combination therapy. The GINA (Global Initiative for Asthma) guidelines position LAMA add-on as a Step 4 to 5 consideration for patients with persistent symptoms or exacerbations on medium-to-high-dose ICS/LABA, which is exactly the population studied in TRIMARAN and TRIGGER.
Trimbow is appropriate to consider for patients who:
- Have documented uncontrolled asthma (frequent symptoms, reliever use, recent exacerbations, or reduced quality of life) despite adherent use of a medium or high-dose ICS/LABA inhaler
- Have experienced at least one moderate or severe exacerbation in the past year
- Have pre-bronchodilator FEV1 below 80% of predicted, indicating measurable airflow limitation
- Are adults (Trimbow is not approved for children)
Trimbow is not appropriate for patients who:
- Need emergency or acute symptom relief (Trimbow is a maintenance inhaler, not a rescue inhaler; always carry albuterol or another SABA)
- Are already using a separate LAMA inhaler (tiotropium, ipratropium, aclidinium, umeclidinium) or another LABA (salmeterol, vilanterol, indacaterol, olodaterol) — combining Trimbow with these creates duplicate drug class exposure with elevated risk of cardiovascular and anticholinergic side effects
- Are children under 18
- Have poorly controlled narrow-angle glaucoma or significant urinary retention from enlarged prostate, as the anticholinergic component can worsen both
Safety: What Patients and Clinicians Need to Know
The safety profile of Trimbow reflects the combined profile of its three components, each of which has decades of clinical experience in other products.
The LABA safety warning
LABAs carry a class-level FDA warning: when used without an ICS, they increase the risk of asthma-related death and hospitalization. This risk is resolved when LABAs are combined with an ICS, as they are in Trimbow. The warning is present on the label but not a contraindication for ICS/LABA combination use. It is the reason that LABAs should never be used as monotherapy in asthma.
Serious risks requiring awareness
- Oral thrush (oropharyngeal candidiasis): The most common ICS-related complication. Rinse and gargle with water after every use, without swallowing. This simple step significantly reduces the risk.
- Adrenal insufficiency: In patients transitioning from oral or systemic corticosteroids to an ICS-containing inhaler, the adrenal glands may not immediately produce sufficient cortisol during physiological stress (illness, surgery, trauma). This risk is real during the transition period and requires gradual steroid tapering under medical supervision.
- Paradoxical bronchospasm: Occasionally, an inhaled medication causes immediate airway tightening rather than opening. If this occurs, stop Trimbow and seek medical care immediately.
- Cardiovascular effects: The beta-agonist and anticholinergic components can increase heart rate and blood pressure. Monitor patients with underlying cardiovascular conditions.
- Glaucoma and urinary retention: The anticholinergic component (glycopyrrolate) increases intraocular pressure and can worsen urinary retention from enlarged prostate. Patients with these conditions need evaluation before starting Trimbow, and regular eye exams are recommended during use.
- Osteoporosis: Long-term ICS use at higher doses contributes to bone density reduction. Assess bone health, particularly in patients with other osteoporosis risk factors.
- Metabolic effects: Elevated blood glucose (particularly in patients with diabetes or at risk for it) and low potassium are possible with LABA therapy.
Common side effects
Bronchitis, upper respiratory infections, hoarseness, mouth and throat discomfort, back pain, and muscle spasms were among the most commonly reported adverse events in clinical trials. Oral hoarseness specifically is related to vocal cord effects of the corticosteroid and can often be reduced by rinsing after use and using a spacer device.
Dosing, Administration, and Storage
| Feature | Details |
|---|---|
| Dose | 2 inhalations twice daily (morning and evening), 4 total puffs per day |
| Available strengths | 86 mcg/4.9 mcg/10.6 mcg (medium) and 172 mcg/4.9 mcg/10.6 mcg (high) per actuation |
| Device type | Pressurized metered-dose inhaler (pMDI) |
| Spacer use | Recommended to improve lung deposition and reduce oropharyngeal deposition |
| Rinse mouth | After every use, with water, without swallowing |
| Storage before first use | Refrigerate at 36°F to 46°F (2°C to 8°C) |
| Storage after first use | Below 77°F (25°C) for a maximum of 2 months; discard after 2 months or when dose counter reaches zero |
| Do not | Freeze; use near heat or open flame; pierce the canister |
| Missed dose | Take as soon as remembered on the same day; skip if it is already the next day; never take more than 4 puffs per day |
If you have previously used a beclomethasone-containing inhaler from a different manufacturer, ask your prescriber about the effective dose. Trimbow’s extrafine formulation may deliver equivalent clinical effect at a lower stated dose than some other BDP-containing products due to improved small airway deposition.
Where Trimbow Fits in the U.S. Asthma Treatment Landscape
Trimbow is the first single-inhaler ICS/LABA/LAMA approved for asthma in the United States. There are existing single-inhaler triple combinations approved for COPD, including Breztri Aerosphere (budesonide/glycopyrrolate/formoterol) and Trelegy Ellipta (fluticasone/umeclidinium/vilanterol), but Trimbow is the first such combination with an asthma-specific approval in the U.S.
Adding a LAMA to ICS/LABA therapy in asthma is already supported by GINA guidelines and the American Thoracic Society/European Respiratory Society guidelines for patients with uncontrolled moderate to severe asthma. What Trimbow provides is the ability to implement this strategy in a single inhaler, which has meaningful practical implications for patients managing multiple devices.
| A note on biologic therapy for severe asthma For patients with severe, refractory asthma and elevated eosinophils or IgE, biologic therapies targeting the type 2 inflammatory pathway are an important consideration alongside or instead of triple inhaled therapy. These include dupilumab (Dupixent), mepolizumab (Nucala), benralizumab (Fasenra), omalizumab (Xolair), and others. These are injected subcutaneous therapies with their own clinical trial programs and indications. Whether triple inhaled therapy or biologic therapy is the appropriate next step for a given patient depends on asthma phenotype, eosinophil count, allergy status, exacerbation history, and other clinical factors best discussed with a pulmonologist or allergist. Trimbow and biologic therapy are not mutually exclusive; some patients may use both. |
|---|
For Patients: What to Discuss With Your Doctor
If your asthma is still not controlled despite consistent use of a combination ICS/LABA inhaler, Trimbow may be worth discussing with your pulmonologist or allergist. The relevant questions are:
- Does my current asthma control score indicate uncontrolled disease despite adherent dual therapy?
- Have I had exacerbations requiring oral steroids or emergency care in the past year?
- Is my pre-bronchodilator FEV1 below normal, suggesting persistent airflow limitation?
- Do I have any contraindications to a LAMA (glaucoma, significant urinary retention, active narrow-angle glaucoma)?
- Am I currently on a separate LAMA or LABA that would need to be discontinued before starting Trimbow?
For general asthma management resources, the American Lung Association and the Asthma and Allergy Foundation of America (AAFA) maintain patient-facing guides on asthma treatment, inhaler technique, and finding specialist care. The GINA Pocket Guide for Patients provides evidence-based guidance on asthma management that patients can review with their healthcare providers.
Sources
FDA approval news: FDA Approves Trimbow (beclomethasone/formoterol/glycopyrrolate) Inhaler for the Maintenance Treatment of Asthma. Drugs.com. May 14, 2026.
Trimbow prescribing information: Trimbow (beclomethasone dipropionate/formoterol fumarate/glycopyrrolate) Prescribing Information. Chiesi USA. 2026.
Drugs.com drug information: Trimbow: Uses, Dosage, Side Effects and Warnings. drugs.com.
TRIMARAN and TRIGGER primary Lancet publication: Virchow JC et al. Single inhaler extrafine triple therapy in uncontrolled asthma (TRIMARAN and TRIGGER): two double-blind, parallel-group, randomised, controlled phase 3 trials. The Lancet. 2019;394(10210):1737-1749. doi:10.1016/S0140-6736(19)32028-9.
TRIMARAN trial registration: NCT02676076. ClinicalTrials.gov.
TRIGGER trial registration: NCT02676089. ClinicalTrials.gov.
Remission-on-treatment analysis: Post-hoc analysis of TRIMARAN and TRIGGER: achieving asthma remission with BDP/FF/GB. Journal of Allergy and Clinical Immunology. 2025.
Subgroup determinants analysis: Determinants of response to inhaled extrafine triple therapy in asthma: analyses of TRIMARAN and TRIGGER. Respiratory Research. 2020. PMC7597025.
Triple therapy review (extrafine formulation): Ridolo E et al. Extrafine formulation of beclomethasone dipropionate/formoterol fumarate/glycopyrronium bromide in the treatment of asthma: a review. Therapeutic Advances in Respiratory Disease. 2025.
Small airways disease in asthma: Small Airways Disease in Asthma. PMC6560600.
LABA FDA safety communication: FDA Drug Safety Communication: New safety requirements for long-acting inhaled asthma medications (LABAs). FDA.gov.
NHLBI asthma overview: Asthma. National Heart, Lung, and Blood Institute.
GINA guidelines: Global Initiative for Asthma (GINA) Reports. ginasthma.org.
Beclomethasone dipropionate: Beclomethasone Dipropionate. StatPearls. NCBI.
Formoterol fumarate: Formoterol Fumarate. StatPearls. NCBI.
Glycopyrrolate/glycopyrronium: Glycopyrrolate. StatPearls. NCBI.
ACQ-7 instrument: Asthma Control Questionnaire validation. PMC4799806.
Patient resources: American Lung Association | Asthma and Allergy Foundation of America | GINA Guidelines
| Disclaimer: Health Evidence Digest provides general information about FDA approvals and health research for educational purposes. This content is not a substitute for professional medical advice, diagnosis, or treatment. Asthma management decisions, including changes to inhaler therapy, should be made in consultation with a qualified pulmonologist, allergist, or primary care provider familiar with your individual asthma history and current treatment. |
|---|
