
| 📌 The essentials On March 26, 2026, the FDA approved KRESLADI (marnetegragene autotemcel, Rocket Pharmaceuticals), the first gene therapy for severe Leukocyte Adhesion Deficiency Type I (LAD-I), indicated specifically for pediatric patients who lack an available HLA-matched sibling donor. The clinical basis: a Phase 1/2 trial (NCT03812263) enrolling 9 children with molecularly confirmed severe LAD-I, showing 100% HSCT-free survival at one year (95% CI 66 to 100; p less than 0.001), 0 graft failures, and sustained neutrophil CD18 expression through median 4.2-year follow-up. Results published in the New England Journal of Medicine. This is an accelerated approval based on biomarker surrogates (CD18 and CD11a expression). Confirmatory post-marketing studies are required. Rare Pediatric Disease Priority Review Voucher granted alongside approval. The boxed warning: lentiviral vector-mediated insertional oncogenesis, requiring long-term post-treatment monitoring. |
|---|
Most children born with severe Leukocyte Adhesion Deficiency Type I do not survive to their second birthday without treatment. Their white blood cells, the immune system’s first responders, lack a critical surface protein called CD18 that allows them to exit the bloodstream and reach the site of infection. Without it, bacteria and fungi go virtually unchallenged. The infections are relentless, poorly responsive to antibiotics, and frequently fatal. Omphalitis, infection of the umbilical stump, is sometimes the first sign, in a newborn just days old.
The only curative option before March 2026 was an allogeneic hematopoietic stem cell transplant from an HLA-matched sibling donor. Most children do not have one. Transplants from mismatched donors carry substantial risks: graft failure, graft-versus-host disease, and transplant-related mortality. Some families faced a situation with no good path forward.
On March 26, 2026, the FDA approved KRESLADI (marnetegragene autotemcel), the first gene therapy for severe LAD-I, indicated specifically for pediatric patients without an available matched sibling donor. The clinical trial behind it enrolled nine children. None had graft failure. All survived. The youngest were infants; all who were enrolled under age one were alive beyond age two. The data was published in the New England Journal of Medicine.
Leukocyte Adhesion Deficiency Type I: What It Is and Why It Kills
LAD-I is caused by biallelic (two-copy) loss-of-function mutations in the ITGB2 gene, which encodes CD18, the beta-2 integrin subunit that pairs with CD11 proteins to form integrin complexes on the surface of leukocytes. These CD11/CD18 complexes, particularly LFA-1, composed of CD11a and CD18, are what allow white blood cells to adhere to the inner walls of blood vessels and migrate through them into infected tissues. Without functional CD18, neutrophils and other leukocytes stay in circulation. They cannot reach wounds. They cannot engulf bacteria at infection sites. Infections that a healthy immune system would clear in days become life-threatening emergencies.
Severe LAD-I is defined by CD18 surface expression below 2% of normal. At this level, even minor infections, a skin abrasion, a gum infection, the umbilical cord stump, can become life-threatening. The infection burden is compounded by delayed wound healing: without leukocyte migration, the inflammatory cascade needed for tissue repair does not function properly.
| How rare is LAD-I? The incidence of LAD-I in the U.S. is estimated at approximately 1 in 100,000 to 1 in 200,000 live births. Roughly two-thirds of affected patients have the severe form. Based on approximately 3.6 million U.S. births per year, that translates to roughly 12 to 24 new cases of severe LAD-I annually in the United States. Global prevalence is higher in regions where consanguineous marriage is more common, as LAD-I is autosomal recessive, meaning a child must inherit one mutated ITGB2 copy from each parent. In some Middle Eastern and South Asian populations, the disease burden is proportionally higher. The ultra-rarity of the disease is part of why gene therapy development has been slow: a clinical trial enrolling 9 patients represents a meaningful fraction of the total global patient population who meet enrollment criteria at any given time. |
|---|
How KRESLADI Works: Lentiviral Gene Correction of the Patient’s Own Stem Cells
KRESLADI is an autologous gene therapy, meaning it is manufactured from the patient’s own cells, corrected in the laboratory, and returned to the same patient. This eliminates the core risk of donor-based transplantation: immune mismatch. There is no foreign tissue to reject, and no graft to mount an attack against the host.
The manufacturing process follows a sequence of steps before the patient receives a single intravenous infusion:
Mobilization and apheresis: The patient receives drugs (G-CSF and/or plerixafor) to mobilize hematopoietic stem cells (HSCs) from the bone marrow into the bloodstream. These CD34+ progenitor cells are then collected by apheresis, a process that filters blood through a machine and harvests stem cells.
Ex vivo gene correction: The harvested CD34+ cells are taken to a manufacturing facility and transduced with a lentiviral vector carrying a functional copy of the ITGB2 gene. The lentiviral vector integrates into the cell genome, providing a permanent genetic correction. A back-up collection of unmodified CD34+ cells is preserved in case engraftment fails.
Myeloablative conditioning: Before infusion, the patient undergoes full myeloablative conditioning, high-dose chemotherapy designed to eliminate the existing defective bone marrow and create space for the corrected cells to engraft. This is a significant clinical intervention and the primary source of short-term treatment-related risk.
Infusion: The gene-corrected cells are infused intravenously in a single dose. They travel to the bone marrow, engraft, and begin producing CD18-expressing neutrophils and other leukocytes, ideally for the patient’s lifetime.
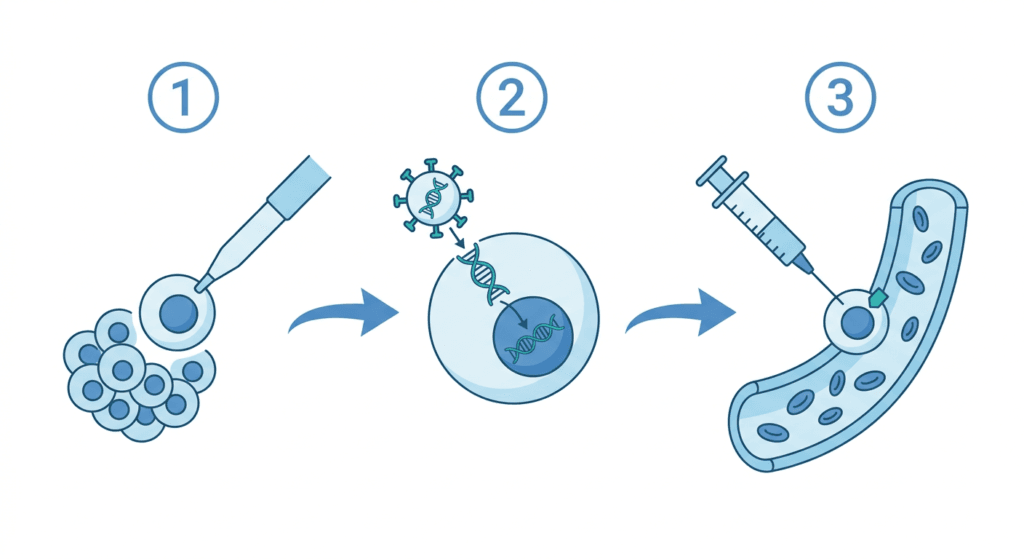
| Lentiviral vectors and insertional oncogenesis: the long-term risk to understand A lentiviral vector works by integrating a copy of the therapeutic gene into the patient’s genome, which is what makes the correction permanent. But integration is not perfectly targeted: the vector can insert near oncogenes (cancer-promoting genes), potentially disrupting their regulation. This risk is called insertional oncogenesis. Early-generation retroviral gene therapies for X-linked SCID caused leukemia in some patients, creating lasting concern about the category. Modern lentiviral vectors like the one used in KRESLADI are self-inactivating (SIN): the viral promoter that could drive oncogene expression is deleted after integration, substantially reducing this risk. The prescribing information for KRESLADI includes a formal warning for lentiviral vector-mediated insertional oncogenesis and requires long-term follow-up monitoring. No cases of insertional oncogenesis were observed in the Phase 1/2 trial, but with a median follow-up of 4.2 years in only 9 patients, long-term surveillance remains a post-marketing requirement. This is not a reason to avoid the therapy in a disease with near-certain early mortality without treatment, but it is a reason for enrolled families to maintain follow-up. |
|---|
The Phase 1/2 Clinical Trial: Nine Patients, Published in NEJM
The trial supporting KRESLADI’s approval (NCT03812263) was an open-label, single-arm, multicenter Phase 1/2 study conducted at leading pediatric immunodeficiency centers including UCLA and Great Ormond Street Hospital in London. It enrolled 9 children with molecularly confirmed severe LAD-I (biallelic ITGB2 mutations, CD18 expression below 2% of normal) who lacked an available HLA-matched sibling donor.
The primary endpoints were biomarker-based: neutrophil CD18 and CD11a surface expression at 12 and 24 months, used as surrogates for restored immune function. Secondary endpoints included safety, engraftment, infection events, and HSCT-free survival. Results were published in the New England Journal of Medicine.
| Outcome | Result |
|---|---|
| Study population | 9 children with severe LAD-I, biallelic ITGB2 mutations, no HLA-matched sibling donor |
| HSCT-free survival at 1 year | 100% (95% CI 66 to 100; p less than 0.001) |
| Graft failures | 0 of 9 |
| Median follow-up | 4.2 years (range 3.6 to 5.7) |
| Neutrophil CD18 expression (Month 12) | Sustained increase in all 9 patients |
| Neutrophil CD18 expression (Month 42) | Sustained in all 7 patients with available data |
| Neutrophil CD11a expression (Month 12) | Median 45% (range 18 to 75) |
| Neutrophil CD11a expression (Month 24) | Median 39% (range 17 to 65) |
| Patients enrolled below age 1 year | All alive beyond 2 years of age |
| Treatment-related serious adverse events | None reported |
| Insertional oncogenesis cases | None observed (ongoing monitoring required) |
Source: Phase 1/2 clinical trial results for marnetegragene autotemcel in LAD-I. New England Journal of Medicine. NCT03812263.
The 100% HSCT-free survival figure warrants careful interpretation alongside the confidence interval (66 to 100%). With only 9 patients, the lower bound of that confidence interval means the true survival rate could theoretically be as low as 66%, which is not a negligible uncertainty. The FDA’s accelerated approval based on biomarker data rather than confirmed long-term clinical outcomes reflects this: the agency considered the surrogate endpoints (CD18 and CD11a expression) sufficiently likely to predict clinical benefit given the biological coherence and the disease’s natural history, while requiring confirmatory data through post-marketing studies.
What the 4.2-year median follow-up confirms is durability of the biomarker response: all 7 patients with data available at month 42 maintained sustained neutrophil CD18 expression. The correction appears stable across the observation period, which is among the longest available for any lentiviral hematopoietic gene therapy in a primary immunodeficiency.
Vinay Prasad, MD, MPH, Chief Medical and Scientific Officer and Director of the FDA Center for Biologics Evaluation and Research, stated at the time of approval that today’s accelerated approval provides a breakthrough treatment for pediatric patients with severe LAD-I, the first FDA-approved gene therapy to treat this disease.
KRESLADI vs. Allogeneic HSCT: Understanding the Comparison
The most meaningful clinical comparison for KRESLADI is not placebo. In a disease with near-certain early mortality, a placebo-controlled trial would be unethical. The relevant comparison is allogeneic hematopoietic stem cell transplantation from an HLA-matched sibling donor, the only prior curative approach.
| Feature | KRESLADI (gene therapy) | Matched sibling HSCT |
|---|---|---|
| Donor required | No, uses patient’s own cells | Yes, HLA-matched sibling |
| Availability | All eligible patients without matched sibling | Approximately 25 to 30% of patients have matched sibling |
| GVHD risk | None (autologous) | Significant, especially with mismatched donors |
| Graft failure risk | 0 of 9 in trial | Higher with mismatched; lower with matched sibling |
| Conditioning required | Yes, full myeloablative | Yes, myeloablative or reduced-intensity |
| Insertional oncogenesis risk | Small but real (lentiviral vector) | None |
| Long-term follow-up data | Median 4.2 years (n=9) | Decades; large registry data available |
| Regulatory status | FDA accelerated approval (March 2026) | Standard curative approach (first-line when donor available) |
The comparison highlights that KRESLADI is approved specifically for the gap population, those without an available HLA-matched sibling donor. It does not replace matched-sibling HSCT, which remains the standard approach when a donor is available. For families without that option, gene therapy now provides a curative path that did not previously exist.
The ongoing clinical evolution of HSCT is also worth noting. Haploidentical transplantation, using a partially matched donor such as a parent, is increasingly feasible through improved graft manipulation techniques and is being evaluated as an alternative for patients without matched siblings. The availability of KRESLADI creates an additional option alongside these evolving transplant approaches, giving clinicians and families more to consider.
Safety: What to Know Before Treatment
No treatment-related serious adverse events were reported in the Phase 1/2 trial, a notable finding given the severity of the conditioning regimen required. Most adverse reactions reflected the expected effects of myeloablative conditioning rather than the gene therapy product itself.
Common adverse reactions in the trial population included: mucositis, upper respiratory tract infection, viral infection, febrile neutropenia, skin lesions, nausea and vomiting, rash, pyrexia, device-related infection, decreased blood counts (hemoglobin, platelets, neutrophils, leukocytes), and elevated liver enzymes (AST, ALT).
The prescribing information includes formal warnings and precautions for:
- Serious infections: susceptibility increases during the myeloablative conditioning period before engraftment is established
- Veno-occlusive disease (hepatic VOD): monitor liver function tests during the first month following infusion
- Neutrophil engraftment failure: defined as failure to achieve absolute neutrophil count of 500 cells per microliter or higher by Day 43; back-up unmodified CD34+ cells are preserved in case rescue is needed
- Delayed platelet engraftment
- Lentiviral vector-mediated insertional oncogenesis: long-term monitoring required
- Hypersensitivity reactions
What Treatment Involves: The Full Patient Journey
For families whose child has been diagnosed with severe LAD-I and lacks an HLA-matched sibling donor, the path to KRESLADI involves a structured sequence of steps that requires specialized care at a qualified treatment center.
Genetic confirmation first
KRESLADI is indicated specifically for patients with severe LAD-I due to biallelic variants in ITGB2 confirmed by molecular testing, with CD18 expression below 2% of normal. Clinical diagnosis alone is not sufficient for eligibility. Genetic testing confirming the ITGB2 mutations is required.
Specialized treatment center
KRESLADI will be administered only at qualified treatment centers with experience in bone marrow transplantation and gene therapy. The manufacturing process, including mobilization, apheresis, ex vivo gene correction, and back-up cell preservation, requires institutional infrastructure that is not available at all pediatric centers. Rocket Pharmaceuticals indicated it plans a measured rollout to ensure quality and safety at launch.
Timing matters
The trial enrolled children as young as infants. Outcomes in LAD-I gene therapy, as in most gene therapies for primary immunodeficiencies, are generally better when treatment is given before significant infection-related damage has accumulated. Families who receive a diagnosis of severe LAD-I should contact a specialized immunodeficiency center promptly to discuss evaluation and next steps, rather than waiting for a clinical crisis.
Regulatory designations
KRESLADI received multiple FDA designations supporting its development:
- Accelerated Approval
- Orphan Drug Designation
- Rare Pediatric Disease Designation
- Breakthrough Therapy Designation
With approval, Rocket Pharmaceuticals received a Rare Pediatric Disease Priority Review Voucher, a transferable regulatory instrument that entitles the holder to priority (6-month) FDA review for a future NDA or BLA. PRVs have historically traded in the $100 to $150 million range. Rocket may use the voucher for a future program or sell it; either way, it represents a financial mechanism designed to incentivize rare pediatric drug development.
For related coverage of other rare pediatric disease FDA approvals in 2026, see our posts on the first gene therapy for genetic deafness approved under the Rare Pediatric Disease PRV program, the UX111 gene therapy BLA for Sanfilippo syndrome now under FDA review, and the approval of navepegritide (YUVIWEL) for achondroplasia in children.
For families whose child has been diagnosed with severe LAD-I or who are under evaluation for a primary immunodeficiency presenting with recurrent severe infections in infancy, the most important first step is early referral to a center with expertise in primary immunodeficiencies and gene therapy. The Immune Deficiency Foundation maintains a directory of immunodeficiency specialists and can connect families with clinical expertise. For questions about KRESLADI specifically, Rocket Pharmaceuticals’ medical affairs team and information on the treatment program are available through their website. The confirmatory post-marketing studies will build the long-term evidence base that the Phase 1/2 trial, while remarkable, cannot yet provide. Families who enroll in long-term follow-up contribute directly to that body of evidence.
Sources
FDA press release: FDA Approves First Gene Therapy for Severe Leukocyte Adhesion Deficiency Type I. FDA.gov. March 26, 2026.
Rocket Pharmaceuticals press release: Rocket Pharmaceuticals Announces FDA Approval of KRESLADI for Pediatric Patients with Severe LAD-I. March 27, 2026. ir.rocketpharma.com.
FDA approval letter: BLA 125806/0 Approval Letter. March 26, 2026.
KRESLADI prescribing information: KRESLADI (marnetegragene autotemcel) Prescribing Information. Rocket Pharmaceuticals; 2026.
NEJM primary publication: Phase 1/2 clinical trial results for marnetegragene autotemcel in LAD-I. New England Journal of Medicine. NCT03812263.
Phase 1/2 trial registration: NCT03812263. Gene Therapy for Leukocyte Adhesion Deficiency Type I. ClinicalTrials.gov.
Pharmacy Times coverage: FDA Approves Marne-Cel, First Stem Cell-Based Gene Therapy for Pediatric Patients With LAD-I. pharmacytimes.com. March 2026.
Rheumatology Advisor: Gene Therapy Kresladi Approved for Severe Leukocyte Adhesion Deficiency-I. rheumatologyadvisor.com. March 2026.
BioPharm International: FDA Approval of Kresladi Expands Gene Therapy in Pediatric Rare Diseases. biopharminternational.com. April 2026.
Disease overview: Novoa EA et al. Leukocyte adhesion deficiency-I: A comprehensive review. J Allergy Clin Immunol Pract. 2018.
LAD-I GARD overview: Leukocyte Adhesion Deficiency Type 1. rarediseases.info.nih.gov.
CD18 biology: CD18 integrin biology. PMC5555401.
Lentiviral vectors: Lentiviral vectors in gene therapy. PMC6563422.
Insertional oncogenesis: Insertional oncogenesis risk in gene therapy. PMC8709598.
Hematopoietic stem cells: Hematopoietic Stem Cells. StatPearls. NCBI.
Myeloablative conditioning: Myeloablative Conditioning. StatPearls. NCBI.
Haploidentical HSCT: Haploidentical Transplantation. PMC7138706.
HLA matching in HSCT: HLA Matching in HSCT. StatPearls. NCBI.
Accelerated approval pathway: Accelerated Approval Program. FDA.gov.
Rare Pediatric Disease PRV: Rare Pediatric Disease Priority Review Voucher Program. FDA.gov.
Patient resources: Immune Deficiency Foundation | NORD: LAD-I | Rocket Pharmaceuticals | ClinicalTrials.gov: LAD-I
| Disclaimer: Health Evidence Digest provides general information about FDA approvals and health research for educational purposes. This content is not a substitute for professional medical advice. Treatment decisions for severe LAD-I should be made in consultation with a board-certified pediatric immunologist or hematologist at a center with expertise in gene therapy and primary immunodeficiencies. KRESLADI received accelerated approval; continued approval may be contingent on confirmatory post-marketing study results. |
|---|