| 📌 The essentials Xeljanz (tofacitinib, Pfizer) was the first-in-class oral JAK inhibitor approved in the world, receiving FDA approval in November 2012 for rheumatoid arthritis. Five indications now: moderate to severe rheumatoid arthritis (RA), psoriatic arthritis (PsA), ulcerative colitis (UC), ankylosing spondylitis (AS), and polyarticular course juvenile idiopathic arthritis (pcJIA). Mechanism: oral small-molecule inhibitor of JAK1, JAK2, JAK3, and to a lesser extent TYK2, blocking the intracellular signaling pathway that dozens of pro-inflammatory cytokines share. Xeljanz generated approximately $625 million in U.S. sales in 2025. The pivotal safety trial: ORAL Surveillance (NCT02092467), a mandated post-marketing Phase 3b/4 trial (n=4,362), found tofacitinib was associated with higher rates of MACE, malignancy, and all-cause mortality compared to TNF inhibitors in RA patients aged 50 and older with cardiovascular risk factors. This resulted in a boxed warning encompassing serious infections, mortality, malignancy, MACE, and thrombosis, and repositioned tofacitinib as a second-line option only after TNF inhibitor failure in RA and PsA. The FDA extended this class-level boxed warning to all JAK inhibitors. Generic entry: the FDA approved the first generic tofacitinib citrate from Ajanta Pharma in August 2025. Full generic competition is now underway. Current Xeljanz list price: approximately $5,000 to $6,000 per month for standard 5 mg twice-daily RA dosing. Expected generic price with multi-source competition: approximately $1,000 to $1,200 per month initially, declining further as competition deepens. The safety warning applies to generic tofacitinib identically to the brand. Generic availability does not make the drug safer for high-cardiovascular-risk patients. |
|---|
| 📚 About this series: the 2026 Loss of Exclusivity Watch This is the final post of HED’s 2026 Loss of Exclusivity series, tracking the ten major drugs losing U.S. exclusivity this year. The full series covers: Xolair (omalizumab) • Pomalyst (pomalidomide) • Opsumit (macitentan) • Januvia/Janumet (sitagliptin) • Simponi (golimumab) • Mavenclad (cladribine) • Gattex (teduglutide) • Trintellix (vortioxetine) • Briviact (brivaracetam) • Xeljanz (tofacitinib). Each post follows the same format: what the drug is and how it works, what the clinical evidence shows, who uses it and why, and what the entrance of competition means for patients, prescribers, and the market. |
|---|
When tofacitinib received FDA approval in November 2012, it was the first oral small-molecule disease-modifying antirheumatic drug approved in the United States in more than a decade and the first-in-class JAK inhibitor anywhere in the world. The approval was the culmination of a genuinely novel drug discovery effort: identifying a target inside the immune cell rather than outside it, designing a molecule small enough to cross cell membranes and block the enzyme, and demonstrating that blocking this enzyme could match the efficacy of the injectable biologics that had dominated inflammatory disease treatment for the preceding decade.
The JAK inhibitor class built on the Xeljanz foundation quickly became one of the most scientifically exciting and clinically contested drug classes in modern medicine. The excitement came from oral administration, rapid onset, broad efficacy across multiple autoimmune diseases, and a reversible mechanism that offered a different risk-benefit profile from continuous biologic immunosuppression. The controversy came from a mandatory post-marketing safety trial, ORAL Surveillance, whose results landed in 2021 and fundamentally reshaped how the entire class is prescribed.
The FDA concluded, based on its completed review of the ORAL Surveillance trial data, that there is an increased risk of serious heart-related events such as heart attack or stroke, cancer, blood clots, and death with tofacitinib, and required a boxed warning for major adverse cardiovascular events, mortality, malignancy, and thrombosis.
Xeljanz generated approximately $625 million in U.S. sales in 2025, down substantially from its peak, with the revenue decline driven by both the prescribing restrictions that followed the safety warning and the early entry of generic competition. The first generic tofacitinib was approved by Ajanta Pharma in August 2025. As of 2026, full generic entry is underway, with prices expected to fall approximately 80%, mirroring the trajectory seen with JAK inhibitor generics in international markets.
This final post in the 2026 LOE series covers tofacitinib’s path from first-in-class JAK inhibitor to a heavily scrutinized drug now entering a generic market, the JAK-STAT pathway it targets, what the ORAL Surveillance data actually says and what it does not say, what it means to prescribe or take tofacitinib in the context of those safety findings, how it compares to the newer and more selective JAK inhibitors that followed it, and what the generic transition means for patients who have been well-controlled on it for years.
What Tofacitinib Treats: Five FDA-Approved Indications
Tofacitinib is FDA-approved for five indications. The breadth of that indication portfolio reflects the JAK-STAT pathway’s central role across multiple immune-mediated inflammatory diseases: the same molecular bottleneck drives inflammation in RA synovium, psoriatic joints, ulcerative colitis mucosa, and the axial skeleton in ankylosing spondylitis.
Rheumatoid arthritis is the primary indication and the one with the deepest evidence base. Since the ORAL Surveillance safety update in December 2021, tofacitinib is specifically indicated for adults with moderate to severe active RA who have had inadequate response or intolerance to one or more TNF blockers. It is no longer a first-line option for RA.
Psoriatic arthritis follows the same post-TNF-failure positioning. Tofacitinib is approved for adults with active PsA who have had inadequate response or intolerance to one or more TNF blockers.
Ulcerative colitis is approved at a higher induction dose (10 mg twice daily for the induction phase, then 5 mg twice daily maintenance, or 10 mg maintenance in patients who do not achieve adequate control), making the UC indication pharmacologically distinct from RA and PsA in terms of dose management.
Polyarticular course juvenile idiopathic arthritis (pcJIA) extends the approved population to children aged 2 years and older, one of the few JAK inhibitor indications in a pediatric population.
Ankylosing spondylitis received its U.S. approval in 2021, positioning tofacitinib as an oral alternative to the TNF inhibitor and IL-17 inhibitor biologics that had previously been standard for biologic-eligible axial spondyloarthritis patients.
The JAK-STAT Pathway: How Tofacitinib Works
To understand tofacitinib’s mechanism and why blocking it suppresses inflammation so broadly, it helps to understand the JAK-STAT signaling pathway, one of the most fundamental communication systems in the immune cell.
When cytokines bind to their receptors on the surface of immune cells, they trigger a cascade of intracellular signaling events. The first step after receptor activation is the cross-phosphorylation of Janus kinase (JAK) proteins, which are bound to the intracellular portion of the cytokine receptor. There are four JAK family members: JAK1, JAK2, JAK3, and TYK2. Different cytokine receptors pair different JAK family members; the specific JAK pair activated determines which downstream signaling molecules are engaged.
Once activated, JAKs phosphorylate STAT proteins, signal transducers and activators of transcription. Phosphorylated STATs form dimers, translocate to the nucleus, and directly activate transcription of genes involved in immune cell proliferation, survival, differentiation, and cytokine production. The result is rapid amplification of the inflammatory signal that began at the cell surface.
Tofacitinib exerts its mechanism by inhibiting intracellular nonreceptor tyrosine kinase JAK enzymes. It inhibits JAK1, JAK2, JAK3, and to a lesser extent TYK2. In cellular settings where JAK kinases signal in pairs, tofacitinib preferentially inhibits signaling by heterodimeric receptors associated with JAK3 and JAK1, with functional selectivity over receptors that signal via pairs of JAK2.
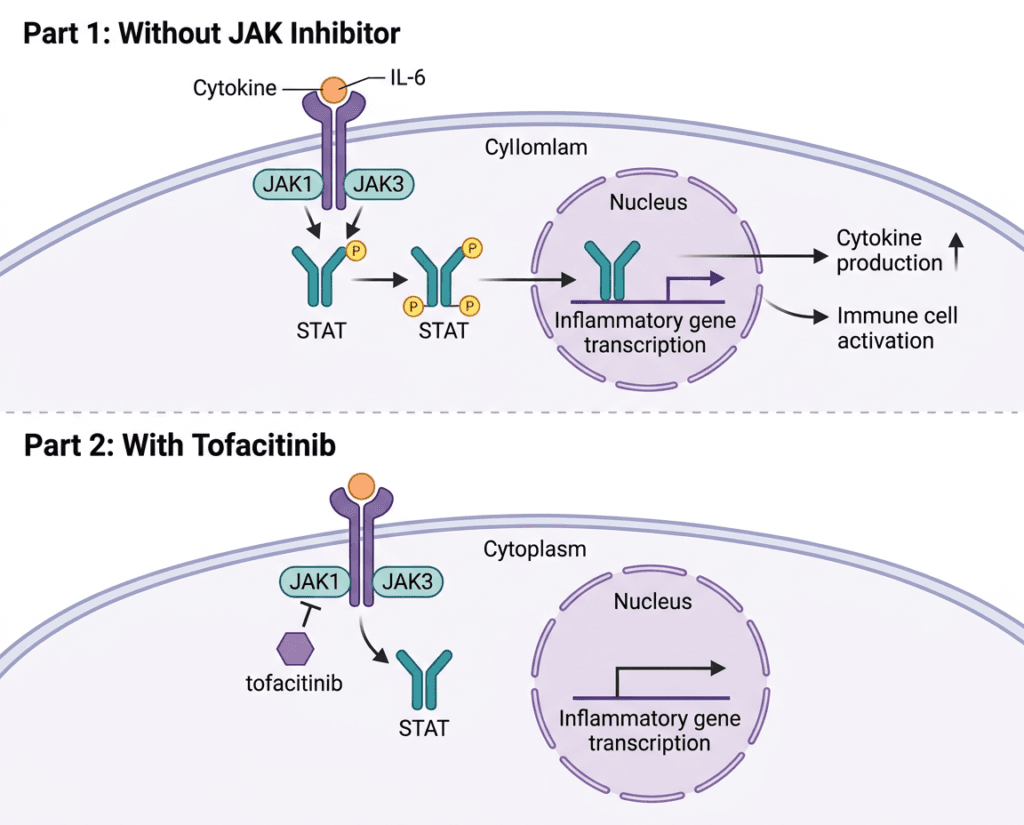
Inhibition of JAK1 and JAK3 blocks signaling through the common gamma chain-containing receptors for several cytokines, including interleukin-2, -4, -7, -9, -15, and -21. These cytokines are integral to lymphocyte activation, development, proliferation, and function. Rather than targeting one cytokine extracellularly the way a biologic monoclonal antibody does, tofacitinib enters the cell and blocks a signaling enzyme that multiple cytokine pathways share. The breadth of that blockade is both the source of its efficacy across multiple diseases and the mechanistic explanation for some of its safety concerns.
| JAK pair inhibited | Cytokines affected | Clinical relevance |
|---|---|---|
| JAK1/JAK3 (primary targets) | IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 | Lymphocyte activation and proliferation; adaptive immunity modulation |
| JAK1/JAK2 | IL-6, IL-10, IL-11, IFN-alpha, IFN-beta | Acute phase response, inflammatory signaling, innate immunity |
| JAK2/TYK2 | IL-12, IL-23 | T-helper cell differentiation; relevant in psoriasis and IBD |
| JAK1/TYK2 | Type I interferons | Antiviral defense; relevant to infection risk |
The ORAL Surveillance Story: What the Data Actually Shows
No discussion of tofacitinib in 2026 can be complete without a thorough and honest account of ORAL Surveillance. It is the single most consequential clinical trial in the history of the JAK inhibitor class and the source of the boxed warning that now governs every tofacitinib prescription.
What the trial was: ORAL Surveillance (NCT02092467) was a Phase 3b/4 open-label, randomized post-marketing safety study mandated by the FDA. The trial enrolled 4,362 patients with moderate to severe rheumatoid arthritis on methotrexate background therapy, all aged 50 years or older and with at least one additional cardiovascular risk factor. Patients were randomized to tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, or a TNF inhibitor (adalimumab in North America, etanercept elsewhere).
What the primary endpoint was: Non-inferiority of tofacitinib to TNF inhibitors for two co-primary endpoints: major adverse cardiovascular events (MACE, defined as cardiovascular death, myocardial infarction, and stroke) and malignancy (excluding non-melanoma skin cancer). The pre-specified non-inferiority margin was an upper bound of 1.8 for the hazard ratio confidence interval.
What the results showed: ORAL Surveillance failed to demonstrate non-inferiority of tofacitinib to TNF inhibitors for both MACE and malignancy. Tofacitinib was associated with numerically higher rates of MACE and malignancy than TNF inhibitors in this high-cardiovascular-risk population.
At the FDA-approved 5 mg twice-daily dose, the number needed to harm was 567 patient-years for MACE and 276 patient-years for malignancy, translating to one additional MACE per approximately 113 patients and one additional cancer per approximately 55 patients treated with tofacitinib instead of a TNF inhibitor over a five-year period. Cancer risk was higher in patients over age 65 (HR 1.70; 95% CI 1.00 to 2.90) than in younger patients (HR 1.36; 95% CI 0.85 to 2.17).
The critical limitations that honest interpretation requires:
First, ORAL Surveillance enrolled a deliberately high-risk population, patients aged 50 and older with established cardiovascular risk factors. The trial lacked a group that was neither a JAK inhibitor nor a TNF inhibitor, meaning it can only compare tofacitinib to TNF blockers, not to placebo or to the underlying disease-related risk. The results therefore quantify the difference in risk between tofacitinib and TNF inhibitors in high-risk patients, not the absolute risk in a typical tofacitinib patient population.
Second, the FDA’s decision to extend the boxed warning to all patients and all JAK inhibitors, not just the high-risk RA population in ORAL Surveillance, was a policy judgment that has been debated in the rheumatology community. Real-world registry data from the Corrona registry, comparing the safety of tofacitinib to other biologics in a broader patient population, found no differences in MACE, serious infection events, malignancy, or death, representing some of the longest-term real-life safety data available for tofacitinib.
Third, the 10 mg twice-daily dose showed more pronounced safety signals than the 5 mg dose, specifically for pulmonary embolism and all-cause mortality. The 10 mg dose is not approved for RA or PsA; its use is limited to the ulcerative colitis induction period. The class warning effectively applied findings from a dose used in RA only in the trial to clinical contexts where that dose would never be used.
The honest clinical summary: ORAL Surveillance demonstrated a real safety signal for cardiovascular events and malignancy in a high-cardiovascular-risk population of RA patients aged 50 and older when comparing tofacitinib to TNF inhibitors. That signal is clinically meaningful for patient selection. It does not characterize the risk-benefit profile in all patients across all indications, and the magnitude of risk in lower-risk patients is substantially less certain.
The Safety Profile: What the Prescribing Information Requires
The Xeljanz prescribing information contains one of the most extensive boxed warning sections in rheumatology, encompassing six distinct categories of serious risk. The infection screening requirements parallel those for the biologic TNF inhibitors discussed in Post 5 of this series on golimumab and the Simponi LOE: tuberculosis screening before initiating, hepatitis B screening, and updated vaccination status are all required. Live vaccines are contraindicated during tofacitinib therapy.
| Safety category | Details | Clinical guidance |
|---|---|---|
| Serious infections (boxed warning) | Increased risk of bacterial, fungal, viral, and opportunistic infections including tuberculosis. Risk is elevated with concomitant immunosuppressives. | Screen for latent TB before initiating. Evaluate for active infection before each refill. Hold tofacitinib during active serious infection. |
| Mortality (boxed warning) | Higher rate of all-cause mortality including sudden cardiovascular death compared to TNF blockers in ORAL Surveillance RA patients. | Use only after failure of TNF inhibitor in RA and PsA. Avoid in patients at high cardiovascular risk unless no suitable alternatives exist. |
| Malignancy (boxed warning) | Higher rates of lymphoma and lung cancer with tofacitinib versus TNF blockers. Risk increased in patients 65 and older, current or past smokers, and those with known malignancy risk factors. | Avoid in patients with known malignancy other than treated non-melanoma skin cancer. Consider alternatives in patients with significant cancer risk factors, especially current or past smokers over 65. |
| MACE (boxed warning) | Higher rate of MACE (cardiovascular death, MI, stroke) versus TNF blockers in ORAL Surveillance, particularly in patients 65 and older, smokers, and those with cardiovascular risk factors. | Use with caution in patients with cardiovascular disease. Avoid in patients at high CV risk unless no suitable alternatives are available. |
| Thrombosis (boxed warning) | Increased incidence of pulmonary embolism, venous thrombosis, and arterial thrombosis, primarily at the 10 mg twice-daily dose. | Use with caution in patients with risk factors for VTE. Promptly evaluate patients reporting signs of DVT or PE. |
| Herpes zoster reactivation | Rates of herpes zoster higher with tofacitinib than with TNF inhibitors. | Ensure zoster vaccination before starting tofacitinib where possible. Monitor during treatment. |
| Hyperlipidemia | Dose-dependent increases in total cholesterol, LDL, and HDL. | Monitor lipid levels 4 to 8 weeks after initiating. Manage dyslipidemia per standard clinical guidelines. |
| Anemia | Hemoglobin decreases observed; avoid initiating in patients with hemoglobin below 9 g/dL. | Monitor CBC during treatment. |
| GI perforations | Cases of GI perforation reported, particularly in patients with Crohn’s disease or diverticulitis. | Use with caution in patients at increased risk for GI perforations. |
| Renal and hepatic impairment | Dose reduction required in moderate renal impairment (eGFR 30 to 60 mL/min) or moderate hepatic impairment. Avoid in severe impairment. | Assess renal and hepatic function at baseline and periodically. |
How Tofacitinib Compares to the Newer JAK Inhibitors
Tofacitinib’s approval opened the door to a JAK inhibitor generation that has continued to evolve. Baricitinib (Olumiant), upadacitinib (Rinvoq), and filgotinib (Jyseleca, approved in Europe but not the U.S.) are more selective for specific JAK isoforms, primarily JAK1, compared to tofacitinib’s broader JAK1/JAK2/JAK3 inhibition.
| Agent | Primary target | Key indications | Selectivity profile | Safety class warning |
|---|---|---|---|---|
| Tofacitinib (Xeljanz) | JAK1/JAK3 | RA, PsA, UC, AS, pcJIA | Broad: inhibits JAK1, 2, 3 | Full boxed warning (class) |
| Baricitinib (Olumiant) | JAK1/JAK2 | RA, alopecia areata, COVID-19 | Preferential JAK1/JAK2 | Full boxed warning (class) |
| Upadacitinib (Rinvoq) | JAK1-selective | RA, PsA, AS, AD, UC, Crohn’s | Highest JAK1 selectivity in class | Full boxed warning (class) |
The theoretical advantage of JAK1 selectivity is that JAK3 inhibition disrupts common gamma chain cytokine signaling (IL-2, IL-7, IL-15) more dramatically and may contribute to a broader immunosuppressive effect that is not necessary for anti-inflammatory benefit in most autoimmune diseases. Whether this translates to meaningful real-world safety differences between agents in the class is still being studied. The FDA extended the class-level boxed warning to all JAK inhibitors in 2021 based on ORAL Surveillance findings, pending further evidence from the class.
For patients: the arrival of generic tofacitinib does not make it the automatic choice over newer, still-branded JAK inhibitors. The prescribing decision should still be driven by individual patient characteristics, cardiovascular risk, prior treatment history, specific indication, and comorbidities, with the rheumatologist or gastroenterologist making a risk-stratified recommendation. What the generic does mean is that tofacitinib becomes far more accessible for patients in whom it is the appropriate choice and for whom cost has been a barrier.
The Generic Entry: What Has Already Happened
The primary composition-of-matter patent for tofacitinib expired in December 2025. In August 2025, the FDA approved the first generic tofacitinib citrate from Ajanta Pharma, Ltd., marking the first generic entry into the Xeljanz market.
As of 2026, full generic entry is underway. Multiple manufacturers have filed ANDAs for generic tofacitinib tablets and extended-release tablets (Xeljanz XR). The immediate-release 5 mg and 10 mg tablets are the highest-volume products; the extended-release 11 mg once-daily tablet has its own separate patent profile and may follow a slightly different generic timeline.
As a small molecule, Xeljanz is far more straightforward to replicate than the monoclonal antibodies used in similar indications. Unlike the biologics covered in this series, golimumab (Simponi) and omalizumab (Xolair), which require complex manufacturing, stability verification, and specialized storage, generic tofacitinib tablets are produced through conventional pharmaceutical chemistry and distributed through standard pharmacy channels. This raises the prospect of rapid substitution, particularly in healthcare systems under cost pressure.
The current Xeljanz list price is approximately $5,000 to $6,000 per month for standard 5 mg twice-daily RA dosing. At approximately 80% price erosion, generic tofacitinib would reach approximately $1,000 to $1,200 per month initially, falling further as competition deepens. For health systems globally where JAK inhibitors have often been reserved for patients with access to payer-negotiated or government-reimbursed pricing, generic availability may meaningfully expand treatment reach.
Pfizer’s response to the LOE includes a branded copay assistance program. As with most specialty drug LOE events in this series, commercial copay assistance for insured patients slows but does not prevent market conversion, with the main beneficiaries of the generic being uninsured patients, Medicare patients, and health systems negotiating formulary contracts.
What Patients Currently on Xeljanz Should Know
If you are currently taking Xeljanz and your disease is well controlled, the generic transition is clinically straightforward. Generic tofacitinib citrate is the same molecule, at the same dose, with the same mechanism. Your disease-modifying benefit, your infection risk, and your safety monitoring requirements are unchanged by the switch from brand to generic.
What does change is cost, in your favor, as formularies transition to preferring the lower-cost generic. Expect formulary notifications about generic tofacitinib in 2026 and into 2027. When that notification arrives, discuss it with your rheumatologist or gastroenterologist at your next visit, not as a cause for alarm, but to confirm your dose and monitoring schedule remain appropriate.
If you are in the high-cardiovascular-risk population that ORAL Surveillance studied, aged 65 or older with cardiovascular risk factors, or a current or past smoker, the prescribing conversation with your rheumatologist should specifically address whether tofacitinib remains the best option for you given the ORAL Surveillance findings, or whether a TNF inhibitor might be more appropriate for your individual risk profile. Generic availability does not change the safety data. It does not make the drug safer for high-risk patients. The boxed warning applies to the generic exactly as it applies to the brand.
For patients newly diagnosed with RA, PsA, or AS: the label now requires demonstrating inadequate response or intolerance to at least one TNF blocker before starting tofacitinib. The generic’s arrival does not change that positioning. It remains a drug for the second-line and later autoimmune treatment setting.
For related HED coverage on other JAK inhibitor approvals and autoimmune disease treatment developments in 2026, see our post on the Simponi (golimumab) LOE and the Immgolis biosimilar litigation and our post on Fasenra (benralizumab) receiving a new indication for hypereosinophilic syndrome.
| 📌 A note on the completed series This post closes out HED’s 2026 Loss of Exclusivity Watch, a 10-post series covering drugs that generated over $17 billion in combined annual U.S. sales now entering the competitive generic and biosimilar market. The series spanned four therapeutic areas and three drug modalities: small molecules (sitagliptin, cladribine, vortioxetine, brivaracetam, tofacitinib, macitentan), a peptide biologic (teduglutide), and injectable biologics (golimumab, omalizumab, pomalidomide). What runs through every post — from Xolair’s interchangeable biosimilar to generic cladribine’s patent invalidation to Xeljanz’s generic entry — is the same fundamental tension in pharmaceutical markets. The periods of exclusivity that fund drug development are real and often necessary. The prices those exclusivity periods produce are frequently out of reach for the patients who need the drugs most. And the generic and biosimilar transitions that eventually bring prices down are complicated, incomplete, and slower in the U.S. than in most other developed health systems. The 2026 patent cliff does not resolve that tension. But for millions of patients currently priced out of Januvia, Trintellix, Mavenclad, Briviact, and Xeljanz, it moves the needle in a meaningful direction. |
|---|
Sources
Xeljanz FDA approval: FDA approves tofacitinib for rheumatoid arthritis. FDA.gov. November 2012.
FDA boxed warning update (December 2021): FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors. FDA.gov. December 2021.
First generic tofacitinib approval (Ajanta Pharma, August 2025): ANDA Drug Approval Database. FDA.gov.
Patent expiry and generic pricing: XELJANZ patent and generic information. DrugPatentWatch. | The next pharma patent cliff: how 2026 to 2032 will reshape revenue. Labiotech. March 2026.
ORAL Surveillance trial registration: NCT02092467. ClinicalTrials.gov.
ORAL Surveillance primary publication: Ytterberg SR et al. Cardiovascular and Cancer Risk with Tofacitinib in Rheumatoid Arthritis. NEJM. 2022;386(4):316–326. doi:10.1056/NEJMoa2109927.
ORAL Surveillance NNH analysis: JAK inhibitors and black box warnings: what is the future for JAK inhibitors? PMC10615860.
Lancet Rheumatology editorial (FDA class warning debate): FDA expands JAK inhibitors warning: going beyond the data? Lancet Rheumatology. 2021.
Corrona registry real-world safety data: Curtis JR et al. Real-world comparative risks of herpes virus infections in tofacitinib and biologic-treated patients with rheumatoid arthritis. Annals of the Rheumatic Diseases. 2021. PMID 34185363.
Tofacitinib mechanism (StatPearls): Tofacitinib. StatPearls. NCBI.
JAK-STAT pathway review: JAK-STAT Signaling Pathway. PMC8440069.
Tofacitinib JAK selectivity in RA: Tofacitinib Suppresses Several JAK-STAT Pathways in RA In Vivo. Frontiers in Immunology. 2021.
JAK inhibitor selectivity comparison: Molecular Modeling Insights into Upadacitinib Selectivity. PMC8778839.
Baricitinib FDA approval: FDA approves baricitinib for moderately to severely active rheumatoid arthritis. FDA.gov.
Upadacitinib FDA approval: FDA approves upadacitinib for moderate to severe rheumatoid arthritis. FDA.gov.
Latent TB screening: Testing for Latent TB Infection. CDC.
Herpes zoster: Herpes Zoster. StatPearls. NCBI.
Xeljanz prescribing information: XELJANZ (tofacitinib) Prescribing Information. Pfizer.
NIAMS disease overviews: Rheumatoid Arthritis | Psoriatic Arthritis | Ankylosing Spondylitis | Juvenile Arthritis
NIDDK ulcerative colitis: Ulcerative Colitis. niddk.nih.gov.
HED internal references: LOE Post 5: Simponi (golimumab) | Fasenra HES approval post
Patient resources: Arthritis Foundation | Crohn’s and Colitis Foundation | Pfizer RxPathways patient assistance | Good Days Patient Assistance
| Disclaimer: Health Evidence Digest provides general information about FDA approvals, loss of exclusivity events, and health research for educational purposes. This content is not a substitute for professional medical advice. Tofacitinib carries a boxed warning for serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis. Decisions about initiating, continuing, or transitioning from brand-name to generic tofacitinib must be made in close collaboration with a board-certified rheumatologist or gastroenterologist who can assess the patient’s individual cardiovascular risk, infection history, and overall benefit-risk profile. Never stop a DMARD without medical guidance. |
|---|