| 📌 The essentials Drug approved: Tecentriq® (atezolizumab) and Tecentriq Hybreza® (atezolizumab and hyaluronidase-tqjs), Genentech/Roche. FDA approval date: May 15, 2026. This is Tecentriq’s eleventh FDA-approved indication. Indication: Adjuvant treatment for adults with muscle-invasive bladder cancer (MIBC) after cystectomy who have circulating tumor DNA molecular residual disease (ctDNA MRD), as determined by an FDA-authorized test. What makes it first-in-class: First ctDNA-guided therapy approval in oncology anywhere in the world. Treatment is triggered by what a serial blood test finds in the weeks after surgery, not by tumor staging alone. Companion diagnostic approved simultaneously: Signatera™ CDx (Natera, Inc.), the personalized ctDNA assay that identifies which patients have molecular residual disease and qualify for adjuvant treatment. Key trial results (IMvigor011, n=250 ctDNA-positive patients): DFS hazard ratio 0.64 (36% reduction in risk of recurrence or death, p less than 0.0001). OS hazard ratio 0.59 (41% reduction in risk of death). ctDNA-negative patients: Those who remained MRD-negative during serial monitoring had 2-year DFS of 88.4% and 2-year OS of 97.1% without receiving any adjuvant treatment. Dosing: Atezolizumab 840 mg IV every 2 weeks, 1200 mg every 3 weeks, or 1680 mg every 4 weeks for up to 1 year, or until recurrence or unacceptable toxicity. Subcutaneous option (Tecentriq Hybreza, 1875 mg every 3 weeks) also approved. |
Nearly half of all patients with muscle-invasive bladder cancer who undergo surgical removal of the bladder will see their cancer return. This is one of the most difficult realities in urological oncology: the patient did everything right, underwent a major operation with significant impact on quality of life, and cancer came back anyway. Often within two years. Often aggressively.
Oncologists have tried for decades to prevent this with adjuvant therapy: chemotherapy or immunotherapy given after surgery to eliminate any cancer cells that surgery may have missed. But a fundamental problem made this approach uncertain. Without a way to know which patients still had cancer cells circulating after surgery, clinicians had to treat everyone at high pathological risk and accept that a large proportion were receiving toxic treatment they did not need, while others with lower-risk staging but hidden disease received nothing.
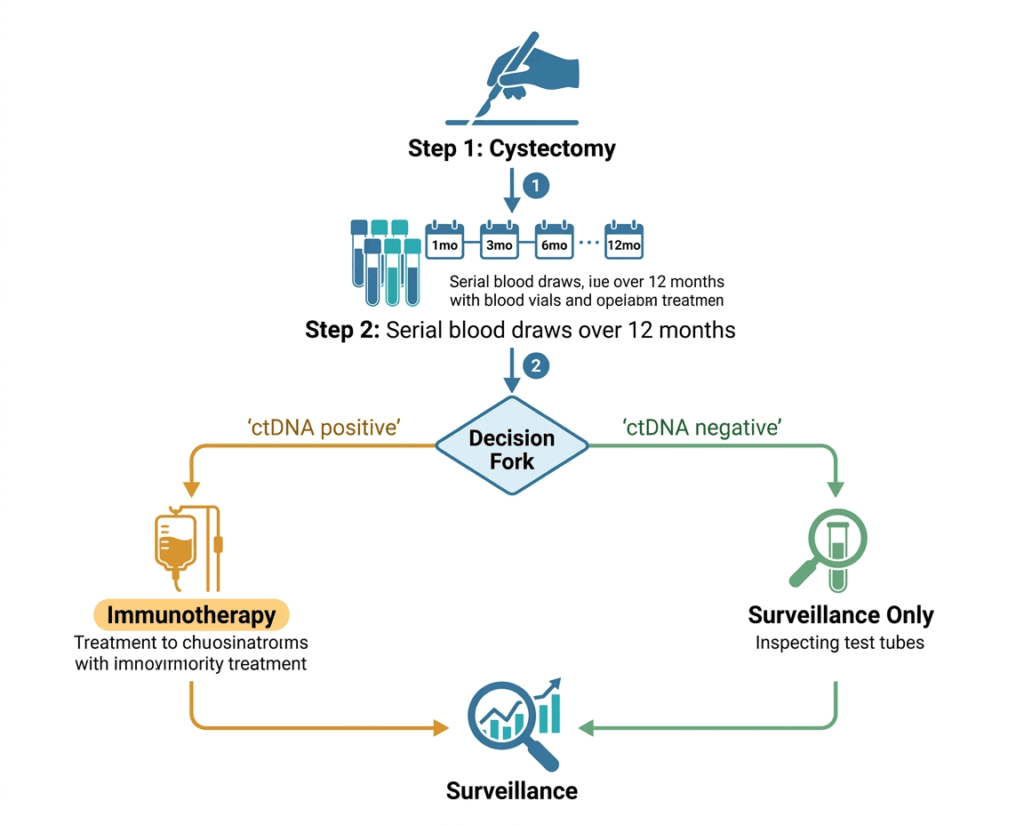
On May 15, 2026, the FDA approved a fundamentally different approach. Using a serial blood test to detect circulating tumor DNA in the weeks after cystectomy, clinicians can now identify exactly which patients still carry molecular evidence of residual cancer. Those who test positive receive adjuvant immunotherapy. Those who remain negative skip it, with data showing they have excellent outcomes without it.
The FDA approval of Tecentriq (atezolizumab) for ctDNA MRD-guided adjuvant treatment of muscle-invasive bladder cancer is the first approval of a ctDNA-guided therapy in oncology history. The companion diagnostic, Natera’s Signatera CDx, was approved the same day. Together they represent what the field has called a new paradigm: treating cancer not based on where it was, but on whether residual evidence of it remains.
Muscle-Invasive Bladder Cancer: Why Adjuvant Therapy Has Been Such a Hard Problem
Bladder cancer is the sixth most common cancer in the United States, with approximately 82,000 new diagnoses annually. The majority are non-muscle-invasive at diagnosis and have a relatively good prognosis with local treatment. Muscle-invasive bladder cancer (MIBC), which has penetrated the muscular wall of the bladder, is diagnosed in roughly 25% of cases and carries a substantially worse prognosis.
Standard treatment for MIBC is radical cystectomy, the surgical removal of the bladder, with or without prior neoadjuvant cisplatin-based chemotherapy. Even with optimal surgical care, nearly half of patients experience disease recurrence within two years, predominantly as distant metastases. At the time progression is detected radiographically, some patients are already too ill to receive further systemic therapy. Earlier intervention, before relapse becomes clinically visible, is the clinical goal.
| Why the prior approach to adjuvant therapy failed in unselected patients The IMvigor010 trial, which directly preceded IMvigor011, tells the story of why patient selection matters. IMvigor010 evaluated adjuvant atezolizumab versus observation in unselected patients with high-risk MIBC after cystectomy. It found no statistically significant improvement in disease-free survival or overall survival with atezolizumab in the overall population. The question that emerged from the failed IMvigor010 result was not whether atezolizumab could work in bladder cancer, but whether the right patients were being selected. A retrospective analysis of the IMvigor010 data identified that approximately 40% of the enrolled patients were ctDNA-positive after surgery. In that ctDNA-positive subgroup, atezolizumab showed a meaningful survival benefit. In ctDNA-negative patients, there was no benefit, because there was no residual cancer for the immune system to target. IMvigor011 was designed prospectively around this insight. Rather than treating all high-risk patients and hoping some would benefit, the study used serial ctDNA testing to identify and enrich the treatment population for the patients most likely to have residual microscopic disease. The result was a positive Phase 3 trial where IMvigor010 had failed, using a different patient selection strategy with the same drug. |
What Circulating Tumor DNA and Molecular Residual Disease Actually Mean
Circulating tumor DNA (ctDNA) is small fragments of DNA shed by cancer cells into the bloodstream. In a patient without cancer, or whose cancer has been completely removed, ctDNA is absent or present at extremely low levels. In a patient with residual cancer cells after surgery, those cells continue to shed tumor-specific DNA into circulation, even before any tumor becomes visible on imaging.
Molecular residual disease (MRD) refers to the presence of detectable cancer at a molecular level, below the threshold of conventional imaging. A patient may have a clear CT scan, clear pathology margins, and no palpable disease, but still have cancer cells circulating or seeded in distant sites at levels that no current imaging can detect. ctDNA testing is the most sensitive available method for detecting this residual disease.
How Signatera™ works
Natera’s Signatera CDx is a personalized, tumor-informed ctDNA assay. This means it is not a generic cancer blood test. Instead, it starts with whole-exome sequencing of the patient’s own tumor tissue to identify up to 16 mutations specific to that individual’s cancer. It then designs a custom PCR panel targeting exactly those mutations to look for them in blood samples over time. This personalization makes Signatera substantially more sensitive than tumor-agnostic ctDNA approaches for detecting low levels of residual disease.
In IMvigor011, serial Signatera testing was performed at multiple time points during the year after cystectomy. Patients who converted from negative to positive during monitoring, or who were positive from the first post-surgical test, were classified as ctDNA MRD-positive and became eligible for the treatment phase. This serial approach identified a 30-day window around each positive test that expanded the period during which patients could be enrolled, rather than requiring a single test result to determine eligibility.
The IMvigor011 Trial: Design and Results
Trial design
IMvigor011 (NCT04660344) is a global Phase 3, randomized, double-blind, placebo-controlled trial. A total of 761 patients with MIBC, no radiographic evidence of disease, and no prior systemic therapy for MIBC were enrolled in the surveillance phase within 6 to 24 weeks of radical cystectomy with lymph node dissection. These patients underwent serial ctDNA monitoring with Signatera for up to one year after surgery.
Of the 761 enrolled, 250 tested ctDNA-positive and entered the treatment phase. They were randomized 2:1 to receive atezolizumab (n=167) or placebo (n=83) every 4 weeks for 12 cycles (approximately one year) or until disease recurrence or unacceptable toxicity. The primary endpoint was investigator-assessed disease-free survival (DFS). Overall survival (OS) was a key secondary endpoint.
Efficacy results
| Endpoint | Atezolizumab (n=167) | Placebo (n=83) |
| Primary: DFS hazard ratio | 0.64 (95% CI 0.44 to 0.93) | Reference |
| Risk reduction in recurrence/death | 36% | N/A |
| DFS p-value | p less than 0.0001 | N/A |
| OS hazard ratio | 0.59 (95% CI 0.37 to 0.94) | Reference |
| Risk reduction in death | 41% | N/A |
| Publication | New England Journal of Medicine (NEJM) | N/A |
| Conference presentation | ESMO 2025 Presidential Symposium (LBA8) | N/A |
Source: Powles T et al. ctDNA-Guided Adjuvant Atezolizumab in Muscle-Invasive Bladder Cancer. NEJM. 2025. doi:10.1056/NEJMoa2511885. Presented at ESMO 2025, LBA8.
A 41% reduction in the risk of death is a substantial survival benefit for an adjuvant setting, where the comparator is placebo (observation) and patients have no detectable disease on imaging. The publication in NEJM and the Presidential Symposium presentation at ESMO 2025 reflect the field’s recognition that this result represents a meaningful advance in bladder cancer care.
The ctDNA-negative population: equally important findings
| Outcome in serial ctDNA-negative patients (n=171, no adjuvant treatment) | Result |
| DFS at 1 year | 95.4% |
| DFS at 2 years | 88.4% |
| OS at 1 year | 100% |
| OS at 2 years | 97.1% |
| Adjuvant treatment received | None |
Source: Natera IMvigor011 topline results. Presented at EAU 2024 and ESMO 2025.
The ctDNA-negative data is as important to the clinical story as the treatment data. Patients who remain serially ctDNA-negative after cystectomy have a 97.1% two-year overall survival without receiving any adjuvant treatment. This means the ctDNA test is not just identifying who needs treatment; it is simultaneously identifying who can safely avoid it. In a disease where adjuvant immunotherapy carries meaningful toxicity and burden, sparing 60 to 70% of post-cystectomy patients from unnecessary treatment is a real clinical benefit.
Why This Approval Matters Beyond Bladder Cancer
The regulatory significance of this approval extends well beyond atezolizumab and bladder cancer. IMvigor011 is the first prospective Phase 3 trial anywhere in oncology to demonstrate that a ctDNA-guided treatment strategy produces statistically significant improvements in both DFS and OS. Every previous ctDNA study in the adjuvant setting had been retrospective or had not yet reported survival outcomes from a randomized controlled trial.
The simultaneous approval of Signatera CDx as a companion diagnostic is the regulatory infrastructure that makes this a replicable model. By pairing a specific ctDNA assay with a specific drug in a specific indication, the FDA has established how ctDNA-guided therapy approvals work. Other drugs and other tumor types in which ctDNA monitoring is being studied now have a regulatory precedent to follow.
ctDNA MRD-guided adjuvant strategies are already being investigated in colon cancer (DYNAMIC trial), lung cancer, breast cancer, and other solid tumors. IMvigor011’s success in bladder cancer, and its accompanying FDA approval, validates the paradigm and accelerates development in those other disease settings.
| What this means for how we think about ‘cancer-free’ after surgery For decades, ‘no evidence of disease’ after cancer surgery meant no visible tumor on imaging and clear margins on pathology. A patient who met those criteria was considered cancer-free and entered a watch-and-wait surveillance period. IMvigor011 establishes that ‘no evidence of disease’ on conventional imaging can coexist with detectable ctDNA in the bloodstream, and the presence of that ctDNA predicts a high probability of recurrence. The approval creates a new, more sensitive definition of post-surgical disease status and a new decision point: not just ‘is imaging clear?’ but ‘is the blood test clear?’ For patients, this changes the conversation after surgery. Serial ctDNA testing is now an FDA-authorized tool to determine whether adjuvant treatment is warranted. A positive result triggers a conversation about immunotherapy. A persistently negative result provides reassurance that the risk of early recurrence is low, and avoids unnecessary treatment. |
Safety: Immune-Mediated Adverse Reactions Are the Primary Consideration
The safety profile of atezolizumab in IMvigor011 was consistent with its established profile across other approved indications. As a PD-L1 immune checkpoint inhibitor, the primary safety concern is immune-mediated adverse reactions: the immune system, once reinvigorated against cancer cells, can also attack healthy tissue.
Immune-mediated adverse reactions can affect virtually any organ system and can range from mild and manageable to severe, life-threatening, or fatal. The systems most commonly involved include the lungs (pneumonitis), liver (hepatitis), intestines (colitis), endocrine glands (thyroid, pituitary, adrenal), kidneys, skin, and nervous system. Most are manageable with corticosteroids and, when necessary, discontinuation of atezolizumab.
Patients receiving atezolizumab after cystectomy should be aware of the immune-mediated adverse reaction spectrum and know to report new or worsening symptoms promptly. Early detection of immune-mediated reactions improves outcomes. The prescribing information provides specific management guidance for each organ system.
What Patients Who Have Had Bladder Cancer Surgery Should Know
- Who is this approval for? Adults with muscle-invasive bladder cancer who have undergone radical cystectomy and who test positive for ctDNA MRD using serial Signatera CDx testing in the year after surgery. It is not for non-muscle-invasive bladder cancer or for patients who have already developed metastatic recurrence.
- When does ctDNA testing happen? Serial testing begins within 6 to 24 weeks of cystectomy and continues for up to one year. Multiple tests are needed because the conversion from negative to positive can happen at any point during the surveillance window. A single negative test at one time point does not provide the same reassurance as persistently negative results across serial tests.
- What happens if I test positive? A positive ctDNA MRD result identifies you as a candidate for adjuvant atezolizumab. Treatment lasts up to one year (approximately 12 cycles). Your oncologist will discuss the benefit-risk balance for your specific situation, including your kidney function, immune history, and other factors that may affect tolerability.
- What happens if I test negative? Persistently ctDNA-negative results during the surveillance period indicate a low risk of early recurrence. The IMvigor011 data shows a 97.1% 2-year overall survival in this group without adjuvant treatment. Regular follow-up imaging continues, but adjuvant immunotherapy is not indicated.
- Is Signatera CDx available now? The Signatera assay has been available commercially for ctDNA testing in several cancer types. The CDx designation for this specific MIBC + atezolizumab indication was granted May 15, 2026. Ask your urologist or oncologist about ordering serial Signatera testing as part of your post-cystectomy surveillance plan.
Resources for bladder cancer patients and caregivers
For patients navigating muscle-invasive bladder cancer treatment and post-surgical surveillance, the Bladder Cancer Advocacy Network (bcan.org) is the leading U.S. patient organization and maintains updated information on approved therapies, clinical trials, and specialist referral resources. For clinicians seeking the full IMvigor011 data, the primary results are published in the New England Journal of Medicine and were presented as a Presidential Symposium abstract at ESMO 2025. Information on Signatera CDx is available through Natera (natera.com).
Sources
FDA approval announcement: FDA approves atezolizumab for adjuvant treatment of muscle invasive bladder cancer in patients with molecular residual disease. FDA.gov. May 15, 2026.
Genentech press release: FDA Approves Genentech’s Tecentriq for Adjuvant Muscle-Invasive Bladder Cancer With ctDNA-Guided Treatment. gene.com. May 15, 2026.
Natera press release (full results): Successful IMvigor011 Trial Achieves 41% Improvement in Overall Survival for Bladder Cancer Patients. natera.com. October 2025.
Primary publication (NEJM): Powles T et al. ctDNA-Guided Adjuvant Atezolizumab in Muscle-Invasive Bladder Cancer. N Engl J Med. 2025. doi:10.1056/NEJMoa2511885.
Renal and Urology News: FDA Approves Atezolizumab for ctDNA-Guided Adjuvant MIBC Treatment. renalandurologynews.com. May 2026.
OncoDaily trial summary: IMvigor011 Trial Reports ctDNA as Predictor of Response to Adjuvant Atezolizumab in Bladder Cancer. oncodaily.com.
Urology Times (ESMO results): ctDNA-guided atezolizumab boosts survival in muscle-invasive bladder cancer. urologytimes.com. February 2026.
Oncology News Central (ESMO presentation): IMvigor011 Data at ESMO 2025 Show ctDNA-Guided Adjuvant Atezolizumab Improves Survival in Bladder Cancer. oncologynewscentral.com.
Targeted Oncology (landmark designation): Landmark IMvigor011 Trial Validates ctDNA-Guided MIBC Therapy. targetedonc.com. March 2026.
IMvigor010 background context: Updated overall survival by circulating tumor DNA status from the phase 3 IMvigor010 trial. Eur Urol. 2024;85(2):114-122.
Patient resources: Bladder Cancer Advocacy Network: bcan.org; Signatera CDx information: natera.com/oncology/signatera/
| Disclaimer: Health Evidence Digest provides general information about FDA approvals and health research for educational purposes. This content is not a substitute for professional medical advice. Treatment decisions for muscle-invasive bladder cancer, including ctDNA testing and adjuvant immunotherapy, should be made in close consultation with a qualified urologic oncologist experienced in bladder cancer management. |