| 📌 What this post covers This is a pipeline post covering early-phase clinical trial data, not an FDA approval. GRWD5769 is investigational: it is not approved for any indication and is not available outside of clinical trials. This post covers Phase 1b data presented at the ASCO 2026 Annual Meeting in an oral session on June 2, 2026. The company: Greywolf Therapeutics, a clinical-stage biotech headquartered in Oxford, UK. The drug: GRWD5769, the world’s first oral inhibitor of ERAP1 (Endoplasmic Reticulum Aminopeptidase 1), a first-in-class mechanism that has never been clinically validated before. The trial: EMITT-1 (NCT06923761), Phase 1b expansion cohorts across 28 centers in Australia, France, Spain, and the United Kingdom. Who was enrolled: patients with six types of advanced solid tumors (lung, liver, bladder, cervical, head and neck, colorectal) who had already developed secondary resistance to prior anti-PD-1 immunotherapy, or microsatellite stable colorectal cancer where checkpoint inhibitors are not effective. These are among the hardest-to-treat populations in oncology. What the data showed: objective response rates of 13 to 36% across all six cohorts; durable clinical benefit rates of 18 to 55%; progression-free survival of 33 weeks in lung cancer and colorectal cancer cohorts; no safety signals at the doses studied. What comes next: Stage 2 cohort expansions are underway to inform a planned randomized Phase 2 study. |
|---|
There is a fundamental problem at the heart of cancer immunotherapy. The immune system works by recognizing abnormal cells as foreign and destroying them. T cells learn to do this by reading the proteins displayed on cell surfaces, antigens that signal “I am not normal.” Cancer immunotherapy, in particular checkpoint inhibitors like the PD-1 drugs that transformed oncology in the 2010s, works by releasing the brakes on T cells so they can attack the cancer they can see.
The problem is that cancer can learn to hide. In a substantial proportion of patients, particularly those whose tumors initially responded to PD-1 inhibition and then progressed, the cancer has found ways to alter or reduce the antigens on its surface, essentially making its cells harder for the immune system to recognize. The T cells that were attacking the cancer are no longer fully activated because the targets they were looking for have changed or disappeared.
This is called secondary anti-PD-1 resistance, and it is one of the most consequential unsolved problems in cancer medicine. For the 30 to 40% of patients who achieve initial benefit from checkpoint inhibitors and then progress, there has been no approved targeted approach to restore immune recognition.
Greywolf Therapeutics, an Oxford-based clinical-stage biotech, has been building toward a mechanistically novel answer: target the enzyme that cancer cells use to edit their surface proteins and alter what the immune system sees. At the 2026 ASCO Annual Meeting, they presented the first clinical evidence that this approach works in human patients.
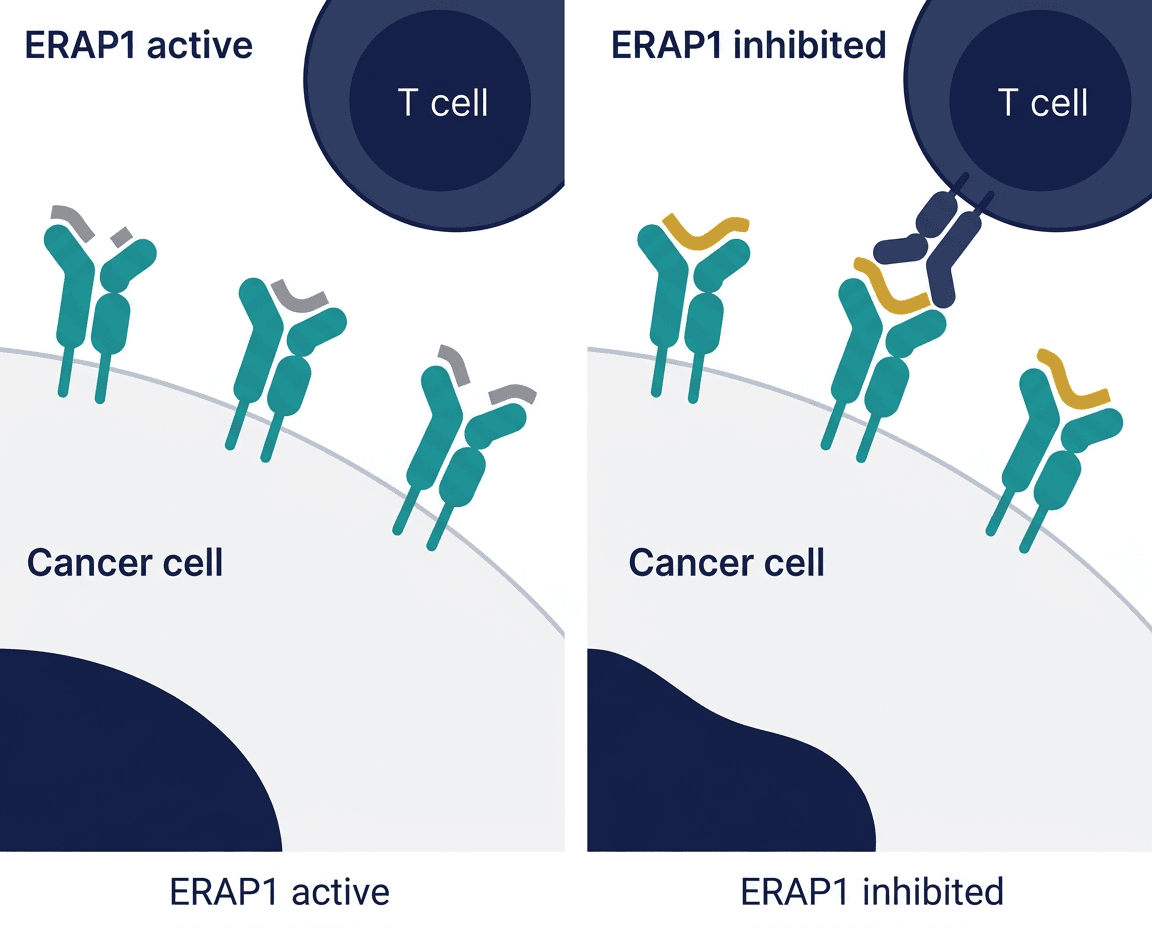
What ERAP1 Is and Why Cancer Uses It to Hide
To understand why ERAP1 inhibition is a new and compelling idea, it helps to understand how cells normally present antigens to the immune system.
Every nucleated cell in the body continuously samples its own intracellular protein content, breaking proteins into short peptide fragments and displaying them on the cell surface via MHC class I (major histocompatibility complex class I) molecules. Cytotoxic T cells patrol for these displayed peptides, looking for fragments that look foreign, viral, bacterial, or the products of oncogenic mutations. When a T cell recognizes a foreign peptide on an MHC-I molecule, it activates and kills the cell displaying it.
The quality and composition of the peptide fragments displayed on MHC-I, collectively called the immunopeptidome, depends critically on which peptides get trimmed to the right length and structure inside the cell. This trimming is performed primarily by ERAP1 (Endoplasmic Reticulum Aminopeptidase 1), an enzyme in the endoplasmic reticulum that trims precursor peptides before they are loaded onto MHC-I for surface display.
ERAP1 is therefore a gatekeeper of antigen presentation. It determines which peptide fragments make it to the cell surface and get displayed to T cells. In many cancers, ERAP1 activity or expression is altered in ways that shift the immunopeptidome toward a less immunogenic profile, reducing the display of peptides that T cells could recognize as foreign. This is one of the mechanisms by which cancers can reduce their immunogenicity over time, evading the T cell attacks that checkpoint inhibitor therapy is trying to mount.
By inhibiting ERAP1, the hypothesis is that you can alter the cancer cell’s antigen presentation, shifting the immunopeptidome back toward a more immunogenic profile, effectively making the cancer visible again to T cells that had become ineffective because their targets were hidden or reduced.
| The cycling dosing strategy: why Greywolf uses an on-off schedule GRWD5769 is not taken continuously. It is administered on a cyclical Q3W (every 3 weeks) on-off schedule in the EMITT-1 trial. This design reflects an important scientific principle: if you continuously inhibit ERAP1 and lock the immunopeptidome into one new configuration, T cells will adapt to that new configuration and may eventually become tolerant to it, producing a new form of exhaustion. By cycling on and off, the drug alternates between two different antigenic repertoires, the ERAP1-active state and the ERAP1-inhibited state, effectively generating a broader range of tumor antigens for T cells to respond to over time. This dynamic approach to antigen presentation is designed to generate more diverse T cell responses and to prevent the chronic antigen exposure that leads to T cell exhaustion, one of the fundamental drivers of immunotherapy resistance. |
|---|
The EMITT-1 Trial: Design and Patient Population
EMITT-1 (NCT06923761) is an adaptive Phase 1/2 global study evaluating GRWD5769 in combination with cemiplimab (Libtayo, Regeneron/Sanofi), an approved anti-PD-1 checkpoint inhibitor. The trial is ongoing at 28 centers across Australia, France, Spain, and the United Kingdom.
The Phase 1b expansion cohorts were designed to enroll patients with specific tumor types and a shared defining characteristic: most had documented secondary resistance to prior anti-PD-1 therapy. These patients had received checkpoint inhibitors, achieved some benefit, and then had their disease progress. For patients in the microsatellite stable colorectal cancer (MSS-CRC) cohort, no prior PD-1 therapy was required because checkpoint inhibitors are not licensed for this indication and are generally ineffective; this represents a population where immunotherapy has never worked.
Six tumor types enrolled:
- Non-small cell lung cancer (NSCLC)
- Urothelial carcinoma (UC, bladder cancer)
- Hepatocellular carcinoma (HCC, liver cancer)
- Microsatellite stable colorectal cancer without liver metastases (MSS-CRC NLM)
- Squamous cell carcinoma of the head and neck (SCCHN)
- Cervical cancer
Patients had a median of 2 prior lines of therapy, confirming this is a heavily pretreated population. 14 evaluable patients were enrolled in five cohorts; 12 were enrolled in the MSS-CRC cohort. The principal investigator is Professor Fiona Thistlethwaite, Consultant Medical Oncologist and Medical Director of the NIHR Manchester Clinical Research Facility at The Christie NHS Foundation Trust.
The ASCO 2026 Data: What Every Cohort Showed
The data table below comes directly from the Greywolf Therapeutics press release accompanying the oral session presentation at ASCO 2026 (Abstract 2500):
| Tumor type | Evaluable patients | Partial responses | ORR | Durable clinical benefit (DCB ≥6 months) | Median PFS |
|---|---|---|---|---|---|
| Urothelial carcinoma (bladder) | 14 | 5 | 36% | 36% | 8 weeks |
| Non-small cell lung cancer | 14 | 3 | 21% | 55% | 33 weeks |
| Hepatocellular carcinoma (liver) | 14 | 2 | 14% | 32% | 16 weeks |
| MSS colorectal cancer (no liver mets) | 12 | 2 | 17% | 51% | 33 weeks |
| Cervical cancer | 14 | 2 | 14% | 18% | 9 weeks |
| Head and neck squamous cell carcinoma | 8 | 1 | 13% | 38% | 14 weeks |
Source: Greywolf Therapeutics ASCO 2026 press release. GlobeNewswire. June 2, 2026. Median follow-up: 8.8 months; data still maturing.
The ORR findings
The objective response rates of 13 to 36% may not look dramatic in absolute terms, but they need to be evaluated against the baseline. These are patients who have already failed anti-PD-1 therapy. When a patient has secondary resistance to a PD-1 inhibitor and is given the same PD-1 inhibitor again, the expected response rate is generally below 5%. Achieving 21% in lung cancer and 36% in bladder cancer in this population represents a meaningful signal that the combination is restoring immune activity against tumors that had become resistant to the mechanism.
The urothelial carcinoma ORR of 36% is the highest single-cohort response rate and is striking in a disease where second-line options after checkpoint inhibitor failure are limited.
The durable clinical benefit findings: the most important numbers
The durable clinical benefit (DCB) rate, defined as complete response, partial response, or stable disease lasting at least 6 months, is arguably the more clinically meaningful metric in a Phase 1b setting. It captures patients who are deriving sustained benefit from the treatment, not just those who had a measurable tumor reduction.
A 55% DCB rate in NSCLC patients with secondary anti-PD-1 resistance means more than half of evaluable lung cancer patients remained progression-free or showed response for at least 6 months. In a setting where treatment options are limited and durable benefit is, in Professor Thistlethwaite’s words, “exceptionally rare,” that is a clinically compelling signal.
The 51% DCB in MSS-CRC is notable for a different reason: microsatellite stable colorectal cancer is one of the cancers most resistant to immunotherapy. Checkpoint inhibitors do not work in this population at standard doses, and the failure of multiple combination approaches in this setting has been a persistent frustration in colorectal cancer oncology. Any 6-month durable benefit rate above 30 to 40% in MSS-CRC is worth attention.
The PFS findings: durability context
The 33-week (approximately 8-month) median PFS in both NSCLC and MSS-CRC cohorts, with follow-up still maturing at 8.8 months, means that at the time of data cutoff, a substantial number of patients were still on treatment and progression-free. This is not a signal of rapid early responses that quickly relapse; it is a signal of sustained disease control in patients who had already exhausted prior options.
What Makes This Mechanism Genuinely New
The immunotherapy field has spent a decade trying to address anti-PD-1 resistance, and most approaches have targeted the same immune checkpoint axes (CTLA-4, LAG-3, TIM-3, TIGIT) or have tried combination chemotherapy-immunotherapy approaches. These have had variable and generally modest success in the secondary resistance setting.
ERAP1 inhibition targets a completely different step in the immunological chain, not the T cell inhibitory signaling that checkpoint inhibitors address, but the upstream process by which the cancer cell prepares and displays the antigens that T cells are supposed to respond to. This is a mechanistically orthogonal approach: rather than removing a brake on T cells, it changes what T cells can see.
The clinical proof-of-mechanism from ASCO 2024 was the first demonstration that a small molecule could pharmacologically modulate the human immunopeptidome in cancer patients. The ASCO 2026 Phase 1b data shows that this modulation, in combination with continued PD-1 blockade, translates into clinical responses and durable disease control in patients who were no longer responding to PD-1 blockade alone.
The combination logic is coherent: re-expose the immune system to a new or altered antigenic repertoire (via ERAP1 inhibition), then allow checkpoint inhibitor therapy to act on the reinvigorated T cell response. The cycling on-off schedule is designed to prevent the new antigenic landscape from itself becoming the source of a new tolerance pattern.
The Safety Signal: Why It Matters at Phase 1b
One of the more notable aspects of the EMITT-1 data is the safety profile. Most adverse events were Grade 1, and no safety signals were identified. This is a Phase 1b combination of a first-in-class oral drug with an approved anti-PD-1 antibody in heavily pretreated cancer patients. The absence of cumulative or overlapping toxicity at efficacious doses is not guaranteed and is practically important: it enables patients to remain on treatment for extended periods, which is what produces the durable disease control signals observed.
The tolerability advantage of an oral delivery mechanism for the ERAP1 inhibitor component, rather than a second intravenous biologic, is also relevant for real-world treatment burden.
What Comes Next
Greywolf has stated that the trial is now advancing into Stage 2 cohort expansions to inform a randomized Phase 2 study. The NSCLC and MSS-CRC cohorts, given their durable clinical benefit signals, are likely to be priority tumor types for expansion and for the Phase 2 design.
The path from Phase 1b to Phase 2 to potential FDA review is measured in years, not months. For the NSCLC setting, which is by far the largest and most commercially significant of the six cohorts, a Phase 2 study with appropriate endpoints (PFS, OS, or biomarker-confirmed ORR) would be required before any regulatory filing could be contemplated. For MSS-CRC, where there are genuinely no effective immunotherapy options and the unmet need is acute, the FDA’s accelerated approval pathway for drugs in serious conditions without adequate alternatives could potentially provide a more compressed timeline if Phase 2 data are compelling.
What EMITT-1 has done is validate the target. ERAP1 inhibition, long recognized as mechanistically compelling in preclinical models, has now produced clinical proof-of-mechanism and Phase 1b efficacy signals across six tumor types. That validation is what enables the field to invest confidently in the next round of trials.
What This Means for Patients With Immunotherapy-Resistant Cancers
For patients with advanced solid tumors who have already received and progressed on PD-1 inhibitor therapy, GRWD5769 is not yet available outside of clinical trials. The EMITT-1 trial continues to enroll across its centers in Australia, France, Spain, and the United Kingdom.
Patients and oncologists who are interested in EMITT-1 enrollment eligibility can check ClinicalTrials.gov (NCT06923761) for current enrollment status and site information. Access to trials at UK centers (notably The Christie in Manchester) or the European sites listed may be relevant for international patients. The Greywolf Therapeutics website at gwt.bio provides additional contact information for clinical trial inquiries.
For patients with MSS-CRC specifically: this is a population with very limited immunotherapy options and where multiple prior combination trials have failed to produce meaningful benefit. The Phase 1b signal in this cohort is the most scientifically surprising finding in the dataset and is likely to generate significant interest from the colorectal oncology community in the Stage 2 expansion.
For related HED coverage of other immunotherapy advances in 2026, see our post on the RP1 (vusolimogene oderparepvec) Complete Response Letter and what the FDA’s evidence standards require for oncolytic immunotherapy approval and our post on Lifyorli (relacorilant) and the novel mechanism of addressing cortisol-mediated chemotherapy resistance in platinum-resistant ovarian cancer.
Sources
Greywolf Therapeutics ASCO 2026 press release (primary source): Greywolf Therapeutics reports durable clinical responses with first-in-class oral ERAP1 inhibitor GRWD5769 across six solid tumor types. GlobeNewswire. June 2, 2026.
BioSpace press release: Greywolf Therapeutics reports durable clinical responses with first-in-class oral ERAP1 inhibitor GRWD5769 across six solid tumor types. biospace.com. June 2, 2026.
European Biotechnology Magazine independent analysis: Greywolf reports early responses with oral ERAP1 inhibitor in solid tumours. european-biotechnology.com. June 2026.
ASCO 2024 proof-of-mechanism press release: Grey Wolf Therapeutics Presents First Clinical Data for GRWD5769 at ASCO 2024. PRNewswire. June 3, 2024.
EMITT-1 trial registration: NCT06923761. ClinicalTrials.gov.
ASCO 2026 Annual Meeting: ASCO Annual Meeting 2026. asco.org.
ERAP1 biology and immunopeptidome: ERAP1 in antigen presentation. PMC7027234.
MHC class I antigen presentation: MHC Class I. StatPearls. NCBI.
Cemiplimab (Libtayo) FDA approvals: Cemiplimab approved indications. FDA.gov.
Greywolf Therapeutics: gwt.bio.
| Disclaimer: Health Evidence Digest provides general information about clinical research and pipeline developments for educational purposes. This content is not a substitute for professional medical advice. GRWD5769 is an investigational drug and is not FDA-approved for any indication. The Phase 1b data presented here represents early-stage evidence; much larger randomized trials are required before any conclusions about efficacy and safety can be drawn for broad clinical use. Patients interested in clinical trial participation should consult with their oncologist and review current eligibility information at ClinicalTrials.gov. |
|---|