Chemotherapy works. For many women with breast cancer, it meaningfully reduces the risk that cancer will return. But chemotherapy also causes real harm: nausea, fatigue, increased infection risk, potential cardiac effects, nerve damage, and in some cases long-term consequences that persist years after treatment ends. For decades, oncologists have known that some women with early-stage breast cancer receive chemotherapy even though their tumor biology would never have threatened them with a recurrence. They endure months of treatment and its side effects for a benefit that, statistically, would not have materialized.
The challenge has always been identifying those women reliably, at the time of diagnosis, before any treatment has started. Existing tools like Oncotype DX and MammaPrint already attempt this, but they require separate molecular testing, come with turnaround times of several days, and cost thousands of dollars. The question the field has been working toward: can AI read a standard pathology slide, combine that with basic clinical data, and produce reliable risk stratification at the point of diagnosis, using materials that already exist?
On May 6, 2026, the FDA cleared ArteraAI Breast (Artera) for exactly that purpose. It is the first FDA-cleared digital pathology-based risk stratification tool for breast cancer. The answer, based on validated clinical trial data, is yes.
| 🔗 Also on HED: AI-Supported Mammography Just Got Its Strongest Evidence Yet This post is part of an ongoing HED series on artificial intelligence in women’s cancer care. Our previous post covered the landmark MASAI trial, which showed AI-supported mammography detected more cancers with no increase in false positives in a 105,000-woman randomized controlled trial. |
Who ArteraAI Breast Is For
ArteraAI Breast is cleared for patients with early-stage, hormone receptor-positive (HR+), HER2-negative invasive breast cancer. This is the most common breast cancer subtype, accounting for approximately 70% of all breast cancer diagnoses. The HR+/HER2- designation means the tumor is driven by estrogen or progesterone signaling and does not overexpress HER2. Standard treatment for early-stage disease in this group includes surgery, radiation, endocrine therapy (hormonal treatment), and, depending on risk, chemotherapy.
The decision about whether to add chemotherapy to endocrine therapy is the key clinical question for most of these patients. Women with clearly high-risk tumors, based on size, lymph node involvement, and grade, typically receive chemotherapy. Women with clearly low-risk disease typically receive endocrine therapy alone. But a substantial middle group sits in ambiguous territory, where the right answer is not obvious from standard pathological features alone. This is precisely the population ArteraAI Breast is designed to help.
How ArteraAI Breast Works
The tool uses multimodal artificial intelligence (MMAI), a term that describes AI systems that combine multiple types of data rather than analyzing a single input. In this case, the two inputs are a digitized histopathology image and patient clinical variables.
The pathology slide input
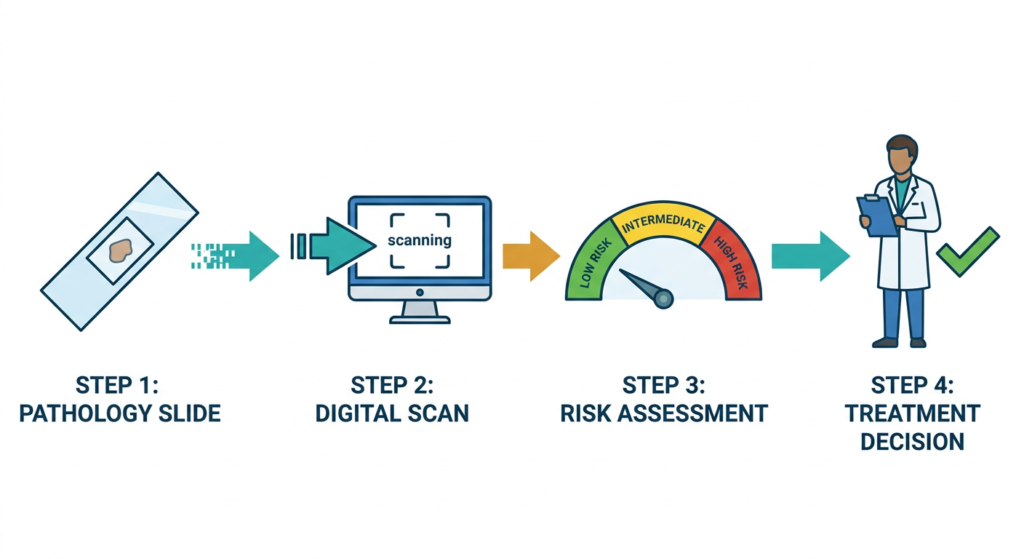
When a breast tumor is surgically removed, tissue samples are processed, embedded in paraffin wax, sliced very thin, stained with standard dyes (hematoxylin and eosin, or H&E), and placed on glass slides. A pathologist reviews these slides under a microscope to assess tumor grade, cell type, and other features. For ArteraAI Breast, the same slides are digitally scanned at high resolution, creating whole-slide images that the AI analyzes. No additional staining, no additional tissue processing, and no additional cost for sample preparation.
The clinical variables input
Alongside the digitized image, the system incorporates standard patient clinical data such as age, tumor size, nodal status, and grade. This multimodal approach allows the AI to recognize patterns across both the visual features of the tumor tissue and the clinical context, producing a composite risk score that neither input alone could generate as accurately.
The output
ArteraAI Breast generates a numerical risk score that provides prognostic information on the likelihood of distant metastasis. Using a predefined risk score cutoff, patients are stratified into low-risk and high-risk groups. Artera reports that results are available within one to two days of receiving the digitized sample, and the test produces no inconclusive results based on insufficient tissue, which is a meaningful practical advantage over some existing molecular assays.
| How does this differ from Oncotype DX and MammaPrint? Oncotype DX (Genomic Health/Exact Sciences) and MammaPrint (Agendia) are the two most widely used molecular risk stratification tests for early-stage HR+/HER2- breast cancer. Both analyze gene expression patterns in tumor tissue and generate recurrence risk scores. Both are validated in large clinical trials (TAILORx for Oncotype DX, MINDACT for MammaPrint) and incorporated into NCCN and ASCO guidelines. The key practical differences with ArteraAI Breast are the input type and the infrastructure required. Oncotype DX and MammaPrint require tumor tissue to be processed with specialized molecular assays, shipped to central laboratories, and analyzed using RNA extraction and gene expression profiling. This adds cost, processing time, and requires specific tissue handling. ArteraAI Breast uses the standard H&E pathology slides that every pathology laboratory already produces as part of routine diagnosis, digitized on equipment increasingly common in pathology labs. ArteraAI Breast does not yet have the decades of clinical validation data behind Oncotype DX and MammaPrint. The tools serve complementary rather than competing roles in the current clinical framework. As the evidence base for ArteraAI grows, the field will develop clearer guidance on how these tools should be used together or sequentially. |
The Clinical Trial Data Behind the Clearance
The FDA clearance is supported by data from two clinical trials, both presented at the 2025 San Antonio Breast Cancer Symposium (SABCS).
ABCSG 8 trial: postmenopausal patients, 10-year outcomes
In a presentation evaluating postmenopausal patients from the ABCSG 8 trial (NCT00291759), the MMAI platform stratified patients into three risk groups with the following 10-year distant metastasis-free survival rates:
| Risk group | 10-year DMFS | Clinical meaning |
| Low risk | Approximately 95% | Very low likelihood of cancer spreading to distant organs within 10 years |
| Intermediate risk | Approximately 89% | Moderate likelihood; additional therapy discussion warranted |
| High risk | Approximately 77% | Substantially elevated risk; chemotherapy benefit more likely to outweigh harm |
NSABP B-20 trial: chemotherapy benefit in high-risk patients
A separate presentation evaluated patients with node-negative, HR-positive disease from the NSABP B-20 trial. In the subset of patients the MMAI tool classified as high-risk, chemotherapy produced a 52% relative decrease in 10-year distant metastasis rates compared with no chemotherapy. This is the predictive component of the tool: not just identifying who has high recurrence risk, but identifying who actually benefits from adding chemotherapy.
The 52% figure is clinically significant. It suggests the AI is not merely sorting patients by overall risk level but identifying the biologically distinct group for whom chemotherapy’s mechanism of action provides substantial additional protection beyond endocrine therapy alone.
Both datasets were presented at SABCS 2025 rather than published in a peer-reviewed journal at the time of FDA clearance. Peer-reviewed publication of the full analyses will be an important milestone for establishing this tool’s position in clinical guidelines.
| “Patients and clinicians need to understand their risks for recurrence and decide which treatments will be the most effective, thereby avoiding both undertreatment and overtreatment.” — Calvin Chao, MD, Vice President of Medical Science, Artera. Medical News Today, May 2026. |
The Bigger Picture: AI Is Changing How Oncologists Make Treatment Decisions
ArteraAI Breast is part of a broader pattern in oncology: artificial intelligence tools are moving from research into regulated clinical practice, with specific cleared or approved uses that change how clinicians gather and act on diagnostic information. The FDA clearance for ArteraAI Breast came in the same month as several other landmark AI decisions in women’s health, reflecting a maturation of the regulatory pathway for these tools.
The clinical and societal significance of AI in this specific context is worth stating plainly. Approximately 300,000 women are diagnosed with breast cancer in the United States each year. A substantial fraction have early-stage HR+/HER2- disease, the exact population for whom the chemotherapy decision is genuinely uncertain. Any tool that reliably identifies the women who can safely avoid chemotherapy reduces harm at scale, not just for individual patients.
The challenge the field now faces is integration. Hospitals need digital pathology scanning infrastructure. Clinicians need to understand what the score means and how to incorporate it alongside existing tools. Guidelines from NCCN, ASCO, and other bodies will need to address how ArteraAI fits alongside Oncotype DX and MammaPrint in clinical decision-making. None of this happens automatically after FDA clearance.
What Patients with Early-Stage HR+/HER2- Breast Cancer Should Know
Is this tool available at my hospital?
ArteraAI Breast received FDA clearance on May 6, 2026. Commercial availability is being rolled out now. Not every hospital or pathology laboratory will have access immediately. Availability depends on whether the institution has digital pathology scanning capability and whether they have contracted with Artera. It is reasonable to ask your oncologist or breast surgeon whether their center uses ArteraAI or a similar digital pathology tool.
Does this replace Oncotype DX or other genomic tests?
Not currently. Oncotype DX and MammaPrint have more extensive published evidence and are incorporated into major clinical guidelines. ArteraAI Breast is a new cleared tool with promising validation data. The two types of tests are based on different biological signals and may provide complementary information. Your oncologist will determine which risk stratification approach is most appropriate for your specific situation.
What does a low-risk result mean in practice?
A low-risk score from ArteraAI Breast indicates that the tumor’s pathological features and your clinical characteristics, as analyzed by the AI, suggest a low probability of distant metastasis. It does not guarantee that cancer will not return. What it does provide is additional evidence that can inform the conversation with your oncologist about whether chemotherapy is likely to offer you a meaningful benefit. That conversation still requires individual clinical judgment, not just a test result.
What limitations exist?
- The supporting data was presented at a conference, not yet published in a peer-reviewed journal. Peer-reviewed publication with full methodology and statistical detail is the standard against which tools are evaluated by guidelines committees. This is expected to follow, and the FDA clearance was granted on the basis of this data, but it is a relevant caveat.
- The tool stratifies into low and high risk, not a single continuous recurrence score. Some other tools provide a continuous score with a range of risk thresholds. The binary or three-tier stratification provides clear decision support but may not capture the full spectrum of risk for every individual patient.
- Long-term prospective data specifically tracking ArteraAI-guided treatment decisions and their outcomes does not yet exist. The existing validation uses retrospective data from prior trials. Prospective evidence that patients guided by ArteraAI scores have better outcomes than those guided by standard assessment alone will take time to accumulate.
The bottom line
For a large number of women with early-stage HR+/HER2- breast cancer, chemotherapy is a treatment they could safely skip. Identifying those women reliably at diagnosis has always been the challenge. ArteraAI Breast is a new, FDA-cleared tool that uses the pathology slide already generated during standard cancer diagnosis to produce a risk score within one to two days, with no additional tissue processing required. The clinical trial data supporting the clearance is promising, particularly the 52% reduction in distant metastasis with chemotherapy in the tool’s high-risk group. The limitations around peer-reviewed publication and prospective outcome data are real and worth tracking. For patients currently navigating a breast cancer diagnosis, the most useful next step is a conversation with a breast oncologist about which risk stratification tools are appropriate for your specific tumor and clinical profile. The National Cancer Institute Cancer Center directory and the Susan G. Komen helpline are strong starting points for connecting with specialized breast oncology care.
Sources
Artera FDA clearance press release: Artera Receives U.S. FDA Clearance for ArteraAI Breast, Expanding Its AI Platform to Breast Cancer. May 6, 2026.
CancerNetwork: FDA Clears AI Stratification Tool in HR+/HER2- Invasive Breast Cancer. CancerNetwork. May 2026.
Femtech Insider: Artera Receives FDA Clearance for AI-Powered Breast Cancer Risk Stratification Tool. Femtech Insider. 2026.
Medical News Today: FDA-cleared AI risk tool could help guide breast cancer therapy. Medical News Today. May 2026.
Medical Device Network: Artera hits US first with pathology-based breast cancer risk tool’s clearance. May 2026.
BusinessWire: Artera Receives U.S. FDA Clearance for ArteraAI Breast. BusinessWire. May 6, 2026.
LabMedica: FDA Clears AI Digital Pathology Tool for Breast Cancer Risk Stratification. LabMedica. 2026.
ABCSG 8 trial: Austrian Breast and Colorectal Cancer Study Group Trial 8 (NCT00291759).
NSABP B-20 trial: National Surgical Adjuvant Breast and Bowel Project B-20. ClinicalTrials.gov.
HED internal post (MASAI): AI-Supported Mammography Just Got Its Strongest Evidence Yet. Health Evidence Digest.
| Disclaimer: Health Evidence Digest provides general information about FDA clearances and health research for educational purposes. This content is not a substitute for professional medical advice. ArteraAI Breast is a risk stratification aid and is not intended to replace clinical judgment. All treatment decisions for breast cancer should be made in consultation with a qualified oncologist. |