It takes an average of 10 to 12 years to bring a new drug from discovery to approval in the United States. A significant portion of that time is not active research. It is waiting. Waiting for data from trial sites to reach sponsors. Waiting for sponsors to analyze and compile that data. Waiting for the FDA to receive a package, assign reviewers, and begin their assessment. Then waiting again, between phases, while the next study design is written and the next application is prepared.
FDA Commissioner Marty Makary put a number on it on April 28, 2026: 45 percent of drug development time is dead time. Not failed experiments or necessary science. Just administrative and logistical lag built into a sequential, phase-based system that has operated largely the same way for 60 years.
On that same date, the FDA announced the launch of real-time clinical trials (RTCT), a new model in which the agency receives safety signals and efficacy data from ongoing trials as they are generated, rather than months or years after the fact. Two cancer drug trials are already live under the program. A broader pilot is scheduled for summer 2026. If the approach works at scale, it could be the most significant structural change to drug development in a generation.
The Problem Being Solved: 60 Years of Sequential Waiting
To understand why real-time clinical trials are significant, it helps to understand exactly how the current system works and where the time goes.
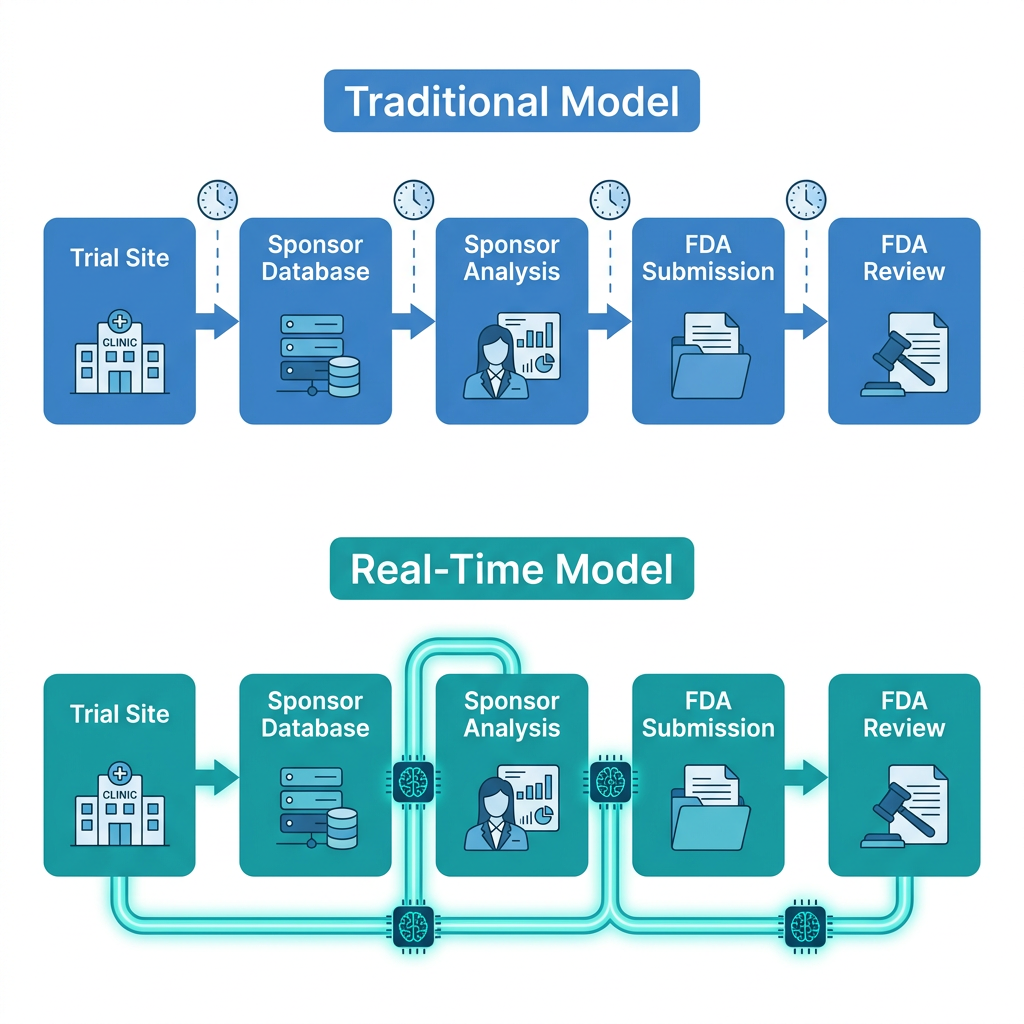
In the traditional model, clinical trial data flows in one direction and at structured intervals. Trial sites collect patient data. That data is periodically uploaded to a sponsor’s database. The sponsor’s biostatistics team analyzes the accumulated data. The analysis is compiled into a formal submission. That submission travels to the FDA. Reviewers are assigned. Review begins. If there is a safety signal, it might be weeks or months old by the time the FDA first sees it. If a Phase 1 trial shows promising efficacy, a new Phase 2 protocol must be written and approved before the next patient is enrolled, with a hiatus in between.
The FDA’s own description of the problem is direct: early-phase trials are characterized by high uncertainty, limited patient populations, and inefficient decision-making processes. Data signals that could immediately inform a dose adjustment, an enrollment modification, or a go/no-go decision sit in a pipeline, aging, while administrative processes catch up.
| What 45% dead time actually means in practice Commissioner Makary’s statement that 45% of drug development time is dead time refers to the gaps and lags built into the sequential phase structure. This includes the interval between Phase 1 completion and Phase 2 initiation, the time between Phase 2 data lock and NDA submission, and the review clock itself. For a drug with a 10-year development timeline, that is roughly 4.5 years that could theoretically be reduced or eliminated if data moved faster and decisions could be made in real time. Even a 20% reduction would translate to approximately 2 years off the development clock for a single drug. The downstream implication for patients is concrete: if promising therapies reach Phase 3 faster, reach the market faster, or are abandoned faster (freeing resources for the next candidate), the aggregate effect on the drug pipeline is substantial. The question is whether real-time data access changes the speed of regulatory decisions, not just the speed of data transfer. |
How Real-Time Clinical Trials Actually Work
The technical infrastructure enabling the RTCT model is built around direct, continuous data connections between trial sites, sponsors, and the FDA, replacing the current batch-submission process with a live data pipeline.
The Paradigm Health SPIRE platform
All trials in the RTCT program use Paradigm Health’s SPIRE platform (Scalable Platform for Integrated Research and Evidence). The platform’s Study Conduct component automates data collection from trial sites and applies AI analysis to identify key safety and efficacy signals. Rather than waiting for the sponsor to run their own analysis and prepare a submission, SPIRE identifies predefined signal thresholds and transmits them to both the sponsor and the FDA as they occur, in days rather than months.
The FDA and each sponsor pre-agree on what constitutes a reportable signal for that specific trial. The criteria are trial-specific and established collaboratively before the trial begins. When the platform detects a signal meeting those criteria, it is transmitted automatically. This means the FDA sees the same data the sponsor sees, at the same time, rather than weeks or months later.
Traditional model versus real-time model
| Step | Traditional model | Real-time model |
| Data collection | Sites collect periodically; upload on schedule | Sites collect continuously; automated upload in near-real time |
| Signal detection | Sponsor runs periodic analyses; prepares formal report | AI platform detects predefined signals immediately |
| FDA access | Months to years after data generated | Days after signal detected; same time as sponsor |
| Safety response | Delayed; based on lagged submissions | Faster; FDA can engage sponsor within days |
| Phase transition | Hiatus between phases; new protocol required | Potential for continuous development; smoother transitions |
| Dose decisions | Based on batch data; slow iteration | Near-real-time signal allows faster dose optimization |
The Two Live Trials: What Is Being Studied and Why Each Was Chosen
TRAVERSE: AstraZeneca, mantle cell lymphoma
The TRAVERSE trial is a Phase 2, multi-site study being conducted by AstraZeneca in patients with treatment-naive mantle cell lymphoma (MCL), an aggressive B-cell blood cancer. The trial evaluates a combination of three targeted agents: acalabrutinib (Calquence, a BTK inhibitor), venetoclax (Venclexta, a BCL-2 inhibitor), and rituximab (an anti-CD20 monoclonal antibody). Sites include The University of Texas MD Anderson Cancer Center and the University of Pennsylvania.
This trial is already live with real-time data flowing to the FDA. Paradigm Health’s platform has received and validated signals from TRAVERSE, establishing that the technical framework works end-to-end in a real clinical trial environment. This proof-of-concept validation is the most concrete achievement in the April 28 announcement: it is not theoretical anymore.
STREAM-SCLC: Amgen, small cell lung cancer
Amgen’s STREAM-SCLC is a Phase 1b study of tarlatamab (Imdelltra), a bispecific T-cell engager targeting DLL3, in patients with limited-stage small cell lung carcinoma. Tarlatamab already has FDA approval for extensive-stage SCLC; this trial is studying the drug in limited-stage disease, which has a more favorable baseline prognosis. Site selection for STREAM-SCLC is still in process, making it slightly behind TRAVERSE in the pilot timeline.
Amgen Chief Medical Officer Paul Burton described the approach at the FDA press conference: the new model sits alongside traditional randomized study approaches rather than replacing them. The STREAM-SCLC trial’s value in the pilot is demonstrating that real-time data transmission works for a Phase 1b study involving a novel mechanism in a disease where dose optimization and safety monitoring are particularly important.
| “For 60 years, we’ve been conducting clinical trials in the same way, where key data signals can take years to reach the FDA. The lag time can delay regulatory decisions unnecessarily and slow down the drug development timeline.” — FDA Commissioner Marty Makary, MD, MPH. April 28, 2026. |
The Broader Pilot: Timeline, Scope, and Who Can Participate
Beyond the two live proof-of-concept trials, the FDA published a Request for Information (RFI) in the Federal Register titled “AI-enabled optimization of early-phase clinical trials pilot program.” This invites sponsors, contract research organizations (CROs), and trial sites to propose studies for inclusion in a broader RTCT program.
| Milestone | Date or detail |
| RFI comment deadline | May 29, 2026 |
| Final selection criteria published | July 2026 |
| Pilot selections complete | August 2026 |
| Pilot program launch | Summer 2026 |
| Platform requirement | All participating trials must use Paradigm Health’s SPIRE platform |
| Priority areas for next cohort | Early-phase oncology, neurology, and rare disease programs |
| Projected benefit | 20 to 40% reduction in overall clinical trial duration |
The RFI specifies that the FDA is looking for sponsors with active early-phase programs in oncology, neurology, and rare diseases. These areas share the characteristic of small patient populations, high medical need, and decision points where real-time data access could most meaningfully accelerate go/no-go decisions. The requirement to use Paradigm Health’s platform creates a standardized data architecture across the pilot, which is essential for the FDA to build operational experience with the model.
What This Means for Patients in Clinical Trials and Patients Waiting for New Treatments
Faster safety response
The most immediate patient-facing benefit of real-time data is faster safety monitoring. In the current model, a safety signal that emerges in week 6 of a trial may not reach the FDA until the next data package is submitted, potentially weeks or months later. Under the RTCT model, that same signal reaches the FDA within days. This means the agency can engage with the sponsor, request a dose modification, or recommend a protocol change much faster than the current system allows. For patients currently enrolled in a trial, this is a direct safety benefit.
Faster development timelines
If the projected 20 to 40% reduction in trial duration holds at scale, the average drug development timeline could compress from 10 to 12 years to somewhere closer to 7 to 9 years. For patients with serious or life-threatening conditions, that difference is not abstract. Every year of acceleration means earlier access to treatments that could change outcomes.
Earlier termination of failing trials
Accelerating development is not only about getting promising drugs to market faster. It also means identifying drugs that are not working or are causing unexpected harm and stopping those trials sooner. In the current model, a drug that is failing may consume years of patient enrollment and sponsor resources before the signal becomes clear enough to act on. Real-time data makes that signal visible earlier.
The Legitimate Questions This Initiative Still Needs to Answer
The RTCT initiative is genuinely promising, and the proof-of-concept success with TRAVERSE is a meaningful milestone. It also raises several questions that the field will need to work through as the program expands.
- Single-platform dependency. Requiring all pilot participants to use Paradigm Health’s SPIRE platform creates a bottleneck of a different kind. If the program expands to dozens or hundreds of trials, a single-vendor infrastructure carries concentration risk. What happens when the platform experiences downtime? Who audits the AI signal detection algorithms for accuracy? These are operational questions the summer 2026 pilot will need to begin answering.
- Data integrity and pre-specified signals. The system works by pre-agreeing on signal definitions before the trial begins. This is scientifically sound but also means the power of real-time data is limited to what sponsors and the FDA anticipated in advance. Unexpected safety signals that do not fit the pre-specified criteria may still be delayed. The governance framework for handling off-protocol signals needs to be explicit.
- Regulatory precedent and legal framework. The traditional clinical trial submission process is embedded in decades of regulation, guidance, and legal precedent. Real-time data sharing between sponsors and the FDA raises questions about whether pre-submission access changes the legal standard for what constitutes a formal submission, how disagreements between the FDA’s real-time assessment and the sponsor’s formal analysis are adjudicated, and what happens to expedited review timelines when continuous data is already available.
- Equity in access to the program. The requirement to use a specific third-party platform adds cost and technical infrastructure requirements that may favor large pharmaceutical companies over smaller sponsors, academic medical centers, and nonprofits. If the program expands with the same single-platform requirement, it risks becoming a tool that primarily accelerates development for companies with the resources to meet the infrastructure bar.
- What “45% dead time” actually includes. Commissioner Makary’s figure is striking, but dead time is not uniformly distributed across drug development. Some of it is genuine administrative lag that faster data pipelines can address. Some of it is necessary scientific deliberation, protocol revision, and peer review that should not be rushed. The 20 to 40% projected time reduction needs to be validated against actual pilot data before it becomes a planning assumption.
Where This Fits in the Broader FDA Modernization Story
The real-time clinical trials initiative did not emerge in isolation. It is part of a pattern of FDA actions in 2025 and 2026 aimed at using artificial intelligence and improved data infrastructure to accelerate drug development without reducing evidence standards. Other pieces of the same picture include the Commissioner’s National Priority Voucher program, which compressed review timelines for priority applications from 10 to 12 months to under 60 days in some cases, and the expansion of the FDA’s AI use in its own review processes.
The RTCT initiative targets a different part of the pipeline than the CNPV program. CNPVs compress the review clock after an NDA is submitted. RTCT aims to compress the development clock before the NDA even exists. Together, they represent a coherent strategy to attack both ends of the 10 to 12 year timeline simultaneously.
What happens next
The May 29, 2026 deadline for RFI comments will be the first public input into how the broader pilot is structured. FDA plans to finalize selection criteria in July and make pilot selections in August. The summer 2026 cohort will be the real test of whether RTCT works at scale across multiple sponsors and disease areas, not just in two carefully chosen proof-of-concept trials.
For patients following drug development in cancer, rare disease, or neurology, the practical upshot is this: if the projected timeline reductions are real, drugs currently in Phase 1 trials could reach Phase 3, or even approval, years sooner than the current system would deliver them. That is a meaningful promise. Whether it holds depends on execution, governance, and the operational questions the next two years of piloting will need to answer. HED will continue tracking the program as the summer 2026 pilot takes shape.
Sources
FDA press announcement: FDA Announces Major Steps to Implement Real-Time Clinical Trials. FDA.gov. April 28, 2026.
HHS announcement: WTAS: FDA Announces Major Steps to Implement Real-Time Clinical Trials. HHS.gov. April 28, 2026.
STAT News (paywalled): FDA testing speedier drug development with real-time clinical trials. STAT News. April 28, 2026. By Lizzy Lawrence.
Fierce Biotech: FDA unveils plan for real-time review of clinical trial data, with AstraZeneca and Amgen already on board. fiercebiotech.com. April 28, 2026.
pharmaphorum: Amgen, AZ will pilot FDA’s real-time clinical trial plan. pharmaphorum.com. April 28, 2026.
Nextgov/FCW: FDA to pilot real-time clinical drug trials through cloud and AI. nextgov.com. April 28, 2026.
Clinical Trials Arena: FDA launches pilot for real-time clinical trials. clinicaltrialsarena.com.
HLTH: FDA Launches Real-Time Clinical Trials Pilot with AstraZeneca and Amgen. hlth.com.
Becaris Publishing: FDA sets out plans for real-time clinical trials, aiming to streamline evidence generation. becarispublishing.com.
Paradigm Health SPIRE platform: Paradigm Health. SPIRE: Scalable Platform for Integrated Research and Evidence. paradigmhealth.ai.
WinBuzzer summary with pilot details: FDA Begins Real-Time AI Trial Pilot with AstraZeneca, Amgen. winbuzzer.com. May 2, 2026.
| Disclaimer: Health Evidence Digest provides general information about FDA regulatory developments and health policy for educational purposes. This content is not a substitute for professional medical advice. The real-time clinical trials program is in an early pilot stage; outcomes, timelines, and program structure are subject to change as the pilot progresses. |