| 📌 The essentials On May 1, 2026, the FDA approved Jakafi XR (ruxolitinib, Incyte), a once-daily extended-release formulation of ruxolitinib, for the same four indications as the original Jakafi: intermediate- or high-risk myelofibrosis (MF) in adults, polycythemia vera (PV) in adults with inadequate response to or intolerance of hydroxyurea, steroid-refractory acute graft-versus-host disease (GVHD) in adults and pediatric patients aged 12 and older, and chronic GVHD after failure of 1 or 2 lines of systemic therapy in adults and pediatric patients aged 12 and older. This is a new dosage form approval, not a new indication. The indications are identical to the original Jakafi. The clinical basis: a randomized, open-label, 2-period, 2-way crossover bioequivalence study (NCT06555081) in 169 healthy adults, showing that 55 mg Jakafi XR once daily is bioequivalent to 25 mg Jakafi twice daily based on steady-state pharmacokinetic measures. Results presented as an ASH 2025 poster (Gong et al. Blood. 2025;146(suppl 1):5045). Safety of Jakafi XR is established from the extensive controlled studies of the original Jakafi across all approved indications: no new safety signals. Available for pharmacy orders beginning May 8, 2026. Critical practical note: Jakafi XR is not a new drug. It delivers the same ruxolitinib molecule at equivalent systemic exposure in a single daily tablet instead of two. Patients who switch should not expect a different clinical effect; the therapeutic benefit comes from the same mechanism and the same total daily drug exposure. |
|---|
Patients with myelofibrosis, polycythemia vera, and graft-versus-host disease have been managing chronic conditions for years on a twice-daily regimen. Twice daily means twice daily: morning and evening, 12 hours apart, building those doses into the rhythm of every single day. For patients who are also managing multiple comorbidities with multiple medications, that twice-daily requirement is one of many adherence demands that can compound into a real burden over time.
On May 1, 2026, the FDA approved Jakafi XR (ruxolitinib), a once-daily extended-release formulation of a drug that has been the standard of care for myelofibrosis and polycythemia vera since 2011 and for steroid-refractory GVHD since 2019 and 2021. The clinical evidence behind the original Jakafi is unchanged. The new formulation simply delivers the same drug at equivalent systemic exposure in a single morning tablet.
This is not a clinical advance in the sense of a new drug with new efficacy data. It is a formulation advance, removing one daily dose from the schedule of patients who are likely to be on this drug for years.
What Ruxolitinib Is and Why These Three Conditions Require Long-Term Treatment
Ruxolitinib is a selective inhibitor of Janus kinase 1 (JAK1) and JAK2, two enzymes that are central to the signaling pathways mediating inflammatory cytokine responses and hematopoietic cell proliferation. JAK1 and JAK2 mediate signaling from multiple cytokine receptors, and their overactivation is the pathological driver in myelofibrosis, polycythemia vera, and GVHD through distinct but related mechanisms.
Understanding why patients take ruxolitinib for extended periods requires understanding each of the conditions it treats.
Myelofibrosis
Myelofibrosis is a myeloproliferative neoplasm in which progressive bone marrow scarring (fibrosis) disrupts normal blood cell production. The fibrosis is driven by inflammatory cytokine overproduction mediated in large part through JAK-STAT pathway activation, often involving somatic mutations in JAK2, CALR, or MPL. The result is progressive anemia, enlargement of the spleen (splenomegaly), constitutional symptoms including debilitating fatigue, night sweats, and weight loss, and over time, the risk of transformation to acute leukemia.
Ruxolitinib was the first approved therapy to substantially reduce splenomegaly and improve constitutional symptoms in intermediate- and high-risk MF. It does not typically reverse or halt fibrosis progression, but it durably reduces the inflammatory burden that drives many of the disease’s most disabling manifestations. Most patients with MF who respond to ruxolitinib continue it indefinitely, as disease symptoms return on discontinuation.
Polycythemia vera
Polycythemia vera (PV) is a chronic myeloproliferative neoplasm characterized by overproduction of red blood cells, often accompanied by elevated white blood cells and platelets, driven almost universally by the JAK2 V617F mutation. The primary risk is thrombosis: the abnormally high blood cell counts and viscosity substantially elevate the risk of stroke, deep vein thrombosis, pulmonary embolism, and other thrombotic events. First-line management involves phlebotomy and aspirin for low-risk patients, and hydroxyurea for high-risk patients. Ruxolitinib is indicated for patients who have had an inadequate response to or are intolerant of hydroxyurea, providing JAK2-targeted suppression of the malignant clone. Again, this is chronic therapy: patients who achieve hematocrit control and symptom relief on ruxolitinib continue it as long as it is effective and tolerated.
Graft-versus-host disease
GVHD is a complication of allogeneic stem cell transplantation in which donor immune cells attack the recipient’s tissues. Acute GVHD typically involves the skin, liver, and gastrointestinal tract; chronic GVHD can affect virtually any organ system and is associated with significant morbidity and mortality. Ruxolitinib, by inhibiting JAK-mediated inflammatory cytokine signaling in donor T cells, reduces the inflammatory cascade driving tissue damage. GVHD patients may require treatment for months to years, again placing this in the category of chronic therapy where dosing schedule matters.
| Why a once-daily formulation matters for chronic blood disease management Patients with MF, PV, and GVHD are frequently older adults with multiple conditions managing multiple daily medications. They may also be managing disease symptoms including fatigue, pain, and cytopenias that affect daily function. In this context, the psychosocial and practical dimension of medication burden is well-documented: adherence to twice-daily regimens is consistently lower than once-daily regimens across all chronic disease areas, even when patients understand the clinical importance of each dose. Studies in conditions from hypertension to HIV have shown that reducing dosing frequency from twice daily to once daily improves adherence by 10 to 20%, with downstream improvements in outcomes that compound over years of treatment. For a drug that works as long as it is taken and loses its effect when stopped, that adherence difference is clinically meaningful. |
|---|
The Approval Basis: What Bioequivalence Means and Why It Is the Right Standard Here
The FDA approved Jakafi XR based on a bioequivalence study, not on a new Phase 3 efficacy trial. This is the correct regulatory approach for a new dosage form of an existing drug with well-established efficacy and safety. Understanding why requires understanding what bioequivalence means and what it does not.
What the bioequivalence study showed
The pivotal study (NCT06555081) was a randomized, open-label, 2-period, 2-way crossover study in 169 healthy adult participants. Each participant received both Jakafi XR 55 mg once daily and Jakafi 25 mg twice daily in random order, with a washout period between. The study measured key pharmacokinetic parameters at steady state: AUC (area under the concentration-time curve, measuring total drug exposure) and Cmax (peak drug concentration).
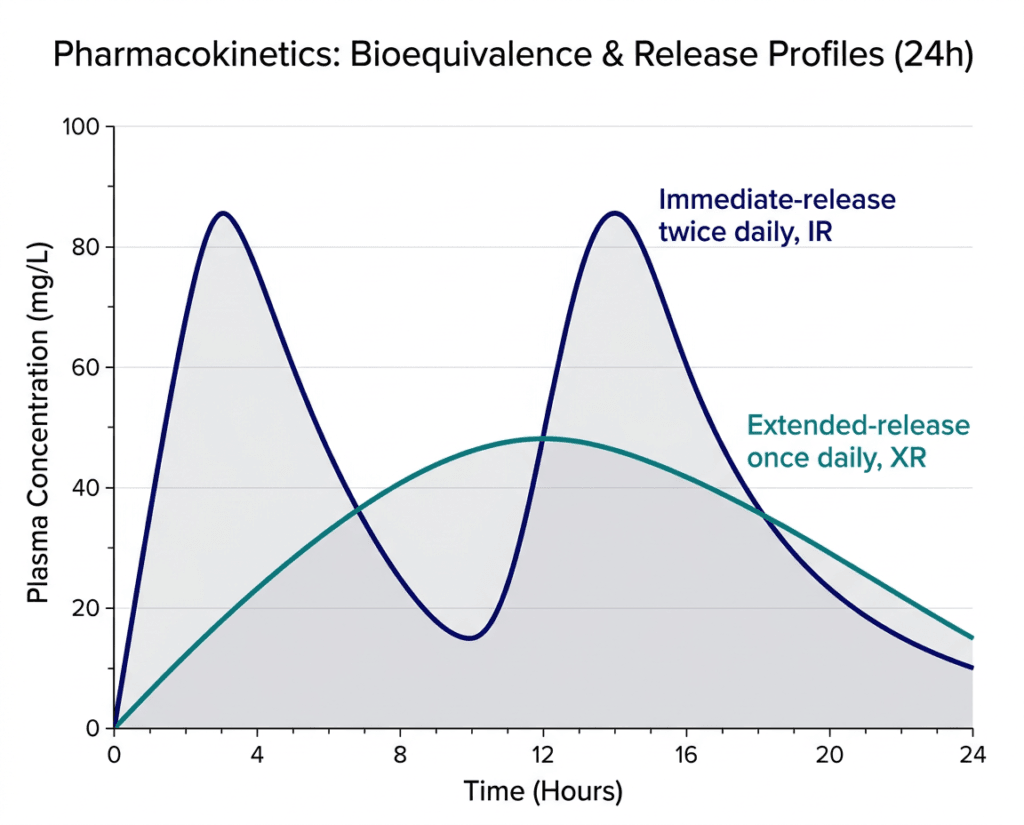
The FDA’s standard for bioequivalence requires that the 90% confidence interval for the ratio of the test (XR) to reference (IR) geometric means for AUC and Cmax falls within 80 to 125%. Jakafi XR met these criteria, confirming that the once-daily extended-release formulation delivers the same total daily ruxolitinib exposure as the twice-daily immediate-release formulation.
Results were presented at the 2025 American Society of Hematology Annual Meeting (ASH 2025) in a poster by Gong et al. (Blood. 2025;146(suppl 1):5045).
| Pharmacokinetic differences between XR and IR: what matters clinically The extended-release mechanism produces a different concentration-time profile than immediate release, even when total daily exposure is equivalent. Jakafi IR produces two peak concentrations per day (one after each dose) with a trough in between. Jakafi XR produces a single, broader, lower peak with more consistent drug levels throughout the 24-hour dosing interval. For JAK inhibition, which requires sustained target coverage rather than intermittent high-concentration peaks, the flatter pharmacokinetic profile of once-daily XR is biologically appropriate. The trough concentrations between IR doses might theoretically allow partial JAK pathway reactivation; the XR formulation maintains steadier JAK inhibition. Whether this translates into any measurable clinical difference in efficacy or tolerability is not yet established from comparative clinical trial data, but the pharmacological rationale supports the XR approach. |
|---|
Why no new Phase 3 trial was required
Ruxolitinib has been studied in multiple large Phase 3 trials across all approved indications, involving thousands of patients with substantial follow-up. The COMFORT-I and COMFORT-II trials established its efficacy in MF; RESPONSE trials established its efficacy in PV; REACH2 and REACH3 established its efficacy in acute and chronic GVHD respectively. Requiring a new Phase 3 trial to approve a formulation change that delivers identical drug exposure would delay access to the more convenient dosing form without generating scientifically useful information. The FDA’s bioequivalence pathway is precisely designed for this scenario.
The Full Jakafi/Jakafi XR Approved Indication Picture
Both Jakafi (original, twice daily) and Jakafi XR (extended release, once daily) now carry the same four indications:
| Indication | Population |
|---|---|
| Intermediate- or high-risk myelofibrosis (primary MF, post-PV MF, post-ET MF) | Adults |
| Polycythemia vera with inadequate response to or intolerance of hydroxyurea | Adults |
| Steroid-refractory acute GVHD | Adults and pediatric patients aged 12 and older |
| Chronic GVHD after failure of 1 or 2 lines of systemic therapy | Adults and pediatric patients aged 12 and older |
Jakafi XR is not approved for use in children with MF or PV; the MF and PV indications are adults only, identical to the original formulation. The GVHD indications cover pediatric patients aged 12 and older with the same weight and age constraints as the original formulation.
Safety: What Applies to Jakafi XR
Because Jakafi XR delivers the same drug at the same systemic exposure, its safety profile is established from the extensive clinical trial database of the original Jakafi across all approved indications. There are no new safety signals from Jakafi XR; the known adverse event profile of ruxolitinib applies in full.
Most important safety considerations for prescribers and patients:
Cytopenias: Thrombocytopenia, anemia, and neutropenia are the most common adverse reactions and can be severe. Dose adjustment or temporary interruption may be required based on complete blood count results. Monitoring is required before initiating and regularly during treatment.
Serious infections: Ruxolitinib increases the risk of serious bacterial, mycobacterial, fungal, and viral infections. Tuberculosis reactivation has been reported. Patients should be evaluated for TB before starting treatment. Progressive multifocal leukoencephalopathy (PML), a serious brain infection caused by JC virus, has been reported rarely with ruxolitinib. Monitoring for signs and symptoms of new neurological symptoms is important.
Herpes zoster: Reactivation of varicella zoster virus (shingles) occurs at higher rates with ruxolitinib than with many other agents. Varicella zoster vaccination before initiating treatment is recommended where possible.
Second primary malignancies: Non-melanoma skin cancers and lymphomas have been reported. Periodic skin examinations are recommended.
Lipid elevations: Increases in cholesterol, LDL, and triglycerides have been reported. Assess lipid levels approximately 8 to 12 weeks after initiating treatment.
Symptom exacerbation on discontinuation: Abrupt discontinuation or dose reduction of ruxolitinib in MF patients can cause rapid return of splenomegaly, constitutional symptoms, and in rare cases a syndrome resembling hemophagocytic lymphohistiocytosis (HLH). Dose tapering is recommended when discontinuation is necessary.
Boxed warning: The FDA requires a boxed warning for thrombosis including fatal cases and for serious infections. These risks apply identically to Jakafi XR as to the original Jakafi.
Dosing: The Key Dose Correspondence Table
Jakafi XR doses correspond to Jakafi (immediate-release) doses at a 2.2:1 ratio reflecting the once-daily to twice-daily conversion. The standard approved correspondence is:
| Jakafi XR (once daily) | Equivalent Jakafi IR dose | Clinical context |
|---|---|---|
| 55 mg once daily | 25 mg twice daily | Standard MF/PV dose; bioequivalence study dose |
| Other XR doses | Corresponding IR twice-daily doses | Per prescribing information; dose adjustments mirror IR guidance |
Administration: Take Jakafi XR orally once daily at approximately the same time each day, with or without food. Swallow tablets whole; do not crush, chew, or split. If a dose is missed, take it as soon as possible on the same day; if the next day has arrived, skip the missed dose.
Switching from Jakafi IR to Jakafi XR: Patients who are stable on Jakafi IR can switch to the corresponding Jakafi XR dose. No dose change is required; the XR dose that matches their current total daily IR exposure should be used. Because switching does not change drug exposure, no transition period or observation period beyond clinical routine is needed.
Practical Implications: Who Benefits Most From the Switch
Not every patient currently on Jakafi needs to switch to Jakafi XR. The clinical effect will be the same. The question is whether once-daily dosing offers enough practical benefit for a given patient to justify the conversation with their hematologist and the pharmacy change.
Most likely to benefit from switching:
- Patients who find twice-daily adherence difficult due to variable daily schedules, work constraints, or fatigue affecting evening dose reliability
- Patients who are managing multiple twice-daily medications and for whom consolidating to once-daily simplifies overall regimen complexity
- Newly initiating patients for whom once-daily is operationally simpler than twice-daily from the outset
- Patients who have expressed frustration with the twice-daily scheduling burden to their hematologist
Unlikely to need switching:
- Patients who are well-established on Jakafi IR with no adherence concerns
- Patients for whom the twice-daily timing provides useful structure or reminders anchored to established daily routines
The key message for patients and clinicians is that Jakafi XR is not a better drug than Jakafi. It is the same drug with a more convenient schedule. In a chronic condition where medication duration is measured in years, a more convenient schedule is a genuine quality-of-life improvement for patients who want it.
For related HED coverage of other formulation advances improving treatment convenience in chronic conditions, see our post on Awiqli, the first once-weekly basal insulin, which similarly reduced the injection frequency from 365 times per year to 52 for appropriate patients with type 2 diabetes.
Sources
FDA approval announcement: FDA approves ruxolitinib extended-release tablets (Jakafi XR). FDA.gov. May 1, 2026.
Incyte press release: Incyte Announces FDA Approval of Jakafi XR (ruxolitinib) Extended-Release Tablets. businesswire.com. May 1, 2026.
Drugs.com approval news: Incyte Announces FDA Approval of Jakafi XR for Myelofibrosis, Polycythemia Vera and GVHD. drugs.com. May 2026.
OncLive clinical coverage: FDA Clears Once-Daily Ruxolitinib Tablets for Myelofibrosis, Polycythemia Vera, and GVHD. onclive.com. May 2026.
Targeted Oncology coverage: FDA Approves Extended-Release Ruxolitinib Once-Daily for MPNs and GVHD. targetedonc.com. May 2026.
Cancer Therapy Advisor clinical review: Jakafi XR Approved for Myelofibrosis, Polycythemia Vera, and GVHD. cancertherapyadvisor.com. May 2026.
CancerNetwork: FDA Approves Ruxolitinib Tablets for Hematologic Malignancies. cancernetwork.com. May 2026.
CURE magazine coverage: FDA Approves Once-Daily Jakafi XR for Myelofibrosis and Other Conditions. curetoday.com. May 2026.
Hematology Advisor: Jakafi XR Approved for Myelofibrosis, Polycythemia Vera, and GVHD. hematologyadvisor.com. May 2026.
ASH 2025 bioequivalence poster: Gong X, Xun Z, Getsy J, McGee R, Mondick J, Punwani N. Bioequivalence of ruxolitinib once-daily extended-release vs twice-daily immediate-release tablets in healthy adults. Blood. 2025;146(suppl 1):5045. doi:10.1182/blood-2025-5045
Bioequivalence study registration: NCT06555081. ClinicalTrials.gov.
Jakafi original FDA approval (2011): FDA approves ruxolitinib for myelofibrosis. FDA.gov. November 2011.
Jakafi prescribing information: Jakafi XR (ruxolitinib) Prescribing Information. Incyte Corporation. 2026.
FDA bioequivalence guidance: Bioequivalence Studies with Pharmacokinetic Endpoints. FDA.gov.
Ruxolitinib mechanism overview: Ruxolitinib. StatPearls. NCBI.
JAK-STAT pathway in MPNs: JAK-STAT Pathway in Myeloproliferative Neoplasms. PMC4207474.
MF cancer overview: Myelofibrosis. American Cancer Society.
PV overview: Polycythemia Vera. NHLBI.
GVHD overview: GVHD Fact Sheet. NCI.
Patient resources: MPN Research Foundation | Leukemia and Lymphoma Society: Myelofibrosis | National MPN Advocacy and Education | Bone Marrow Transplant Info Network
| Disclaimer: Health Evidence Digest provides general information about FDA approvals and health research for educational purposes. This content is not a substitute for professional medical advice, diagnosis, or treatment. Decisions about switching from Jakafi to Jakafi XR, or initiating ruxolitinib therapy in any approved indication, should be made in consultation with a qualified hematologist familiar with the patient’s full clinical history, current blood counts, and concurrent medications. Do not discontinue or change ruxolitinib doses without medical guidance. |
|---|