Uterine cancer is the most common gynecologic malignancy in the United States. This year, an estimated 68,270 women will receive a new diagnosis of endometrial carcinoma, the most common type. Death rates from the disease have been rising steadily since 1997, and the gap between early-stage survival (up to 95%) and late-stage survival (below 20%) is among the widest of any cancer. The difference between those two outcomes often comes down to whether the cancer was caught before it spread.
Unlike cervical cancer, which has a well-established routine screening test (the Pap smear), endometrial cancer has no recommended population-level screening for average-risk women. Diagnosis depends on evaluating symptoms, primarily abnormal uterine bleeding, and performing an endometrial biopsy when warranted. The current standard device for that biopsy is the Pipelle sampler, a thin plastic catheter that has not fundamentally changed in decades.
On April 22, 2026, Utepreva LLC announced FDA 510(k) clearance for the Utepreva Endometrial Sampler, a redesigned single-use device that combines three collection mechanisms in one instrument and supports cytologic, histopathologic, and molecular testing from a single sample. The company says the procedure takes about 20 seconds and requires no dilation, sedation, or operating room. Providers can order it beginning in October 2026.
Endometrial Cancer: The Disease Most Women Know Least About
The endometrium is the inner lining of the uterus. Each month during a woman’s reproductive years, it thickens in preparation for a potential pregnancy and sheds if conception does not occur. When cells in this lining undergo malignant transformation, the result is endometrial carcinoma, the most common uterine cancer by far.
Most endometrial cancers are diagnosed because a woman reports abnormal uterine bleeding to her gynecologist. In postmenopausal women, any vaginal bleeding is considered abnormal and warrants evaluation. Because this symptom tends to appear when the cancer is still localized to the uterus, endometrial cancer is frequently caught at stage I, when surgical removal is usually curative. The problem is the proportion of women who either dismiss the bleeding, delay seeking care, or are told it is nothing to worry about before receiving a proper evaluation.
| The racial disparity in endometrial cancer outcomes Non-Hispanic Black women have the highest mortality rate from uterine cancer of any racial or ethnic group in the United States, and the gap has been widening. Black women are more likely to be diagnosed with aggressive non-endometrioid histologic subtypes (Type II tumors), which account for a disproportionate share of deaths despite representing a minority of total cases. Barriers to timely evaluation, lower rates of specialist access, and differences in tumor biology all contribute. Any advance in detection infrastructure that makes endometrial evaluation faster, less costly, and more accessible in office-based settings has equity implications as well as clinical ones. |
Who Is at Risk: A Practical Guide to Endometrial Cancer Risk Factors
Most endometrial cancer risk comes down to one underlying mechanism: prolonged exposure of the uterine lining to estrogen without the counterbalancing effect of progesterone. Conditions that increase this “unopposed estrogen” exposure elevate endometrial cancer risk. Here is what that looks like in practice.
| Risk factor | What it means clinically |
| Postmenopausal bleeding | The most actionable symptom. Any vaginal bleeding after menopause requires evaluation. This alone is an indication for endometrial sampling regardless of other risk factors. |
| Obesity (BMI 30 or above) | The strongest modifiable risk factor. Excess body fat increases peripheral conversion of androgens to estrogen. Nearly 70% of early-stage endometrial cancer patients are obese. |
| Estrogen therapy without progestogen | Women with a uterus taking estrogen-only hormone therapy have significantly elevated risk. Combination (estrogen plus progestogen) therapy does not carry the same risk. |
| Tamoxifen use | Tamoxifen, used in breast cancer treatment and prevention, acts as an estrogen agonist in the uterus even while blocking estrogen in breast tissue. Women on tamoxifen who develop any abnormal uterine bleeding should be evaluated promptly. |
| Lynch syndrome (hereditary) | Lynch syndrome carriers have a lifetime endometrial cancer risk of 13 to 60%, depending on the gene mutation (MLH1, MSH2, MSH6, PMS2). This is often higher than their colorectal cancer risk. All women with Lynch syndrome should discuss surveillance with their gynecologist. |
| Nulliparity and late menopause | Women who have never been pregnant and those who experienced menopause after age 55 have had longer cumulative estrogen exposure. |
| Diabetes and metabolic syndrome | Hyperinsulinemia and insulin resistance promote endometrial cell proliferation independently of estrogen levels. |
| Age 50 to 70 | Incidence peaks in this age range, coinciding with the postmenopausal transition and its associated hormonal changes. |
| Family history | A first-degree relative with endometrial or colorectal cancer warrants discussion about Lynch syndrome testing even if the patient does not meet formal criteria. |
Sources: AAFP (Am Fam Physician. 2025;111(6):526-531), StatPearls Endometrial Cancer, StatPearls Postmenopausal Bleeding.
How Endometrial Cancer Is Currently Diagnosed
When a woman presents with postmenopausal bleeding or other concerning symptoms, the standard evaluation pathway begins with a pelvic examination and often a transvaginal ultrasound to measure endometrial thickness. An endometrial stripe of more than 4 mm in a postmenopausal woman with bleeding is an indication for tissue sampling. Even a stripe below that threshold does not rule out malignancy if bleeding is persistent.
Tissue sampling is performed with an endometrial sampler inserted through the cervix into the uterine cavity. The Pipelle sampler, introduced in the 1980s, remains the most commonly used device in the United States. It is a thin plastic catheter that uses a retractable piston to create suction and aspirate endometrial tissue. It works well in straightforward cases but has documented limitations: it samples a fraction of the uterine cavity, may produce insufficient tissue in certain uterine configurations, and cannot collect the range of sample types (cytologic, histopathologic, molecular) from a single pass that modern testing increasingly requires.
When the Pipelle yields insufficient tissue, or when a focal lesion is suspected, the next step is dilation and curettage (D&C) under sedation or general anesthesia, with or without hysteroscopy. This requires operating room resources, carries anesthesia risk, and adds cost and scheduling delay.
| 🔗 Also on HED: Vaginal Estrogen Safety in Endometrial Cancer Survivors Our previous post covered a landmark 2,824-patient matched cohort study and the FDA’s February 2026 removal of the boxed warning for vaginal estrogen in endometrial cancer survivors. Relevant background for anyone following uterine cancer care https://healthevidencedigest.com/vaginal-estrogen-therapy-not-linked-to-cancer-recurrence-in-younger-survivors-of-endometrial-cancer/ |
What the Utepreva Endometrial Sampler Is and How It Works
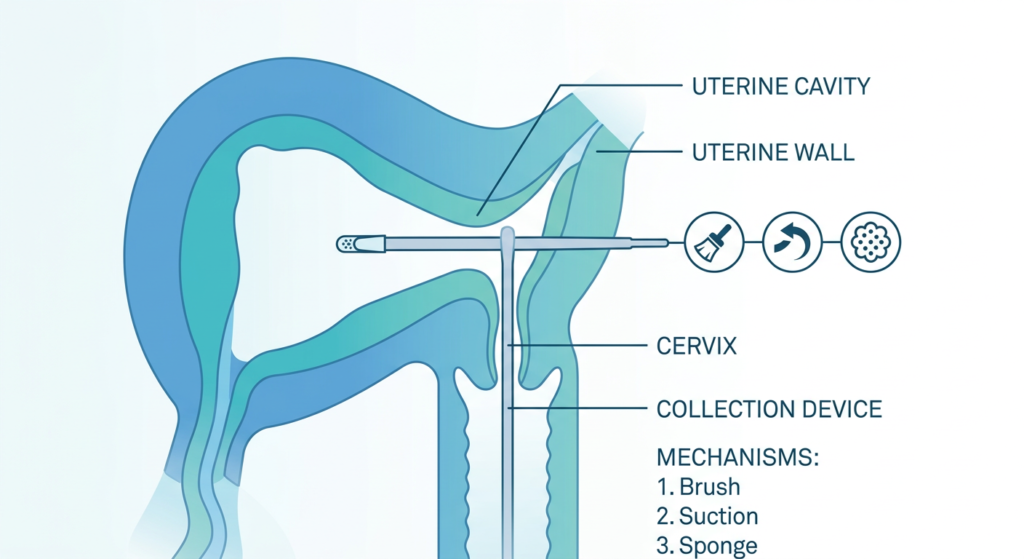
The Utepreva Endometrial Sampler (510(k) clearance K240595) is a single-use, patented device that combines three distinct tissue collection mechanisms in one instrument, intended to be performed as a single-pass sampling procedure.
The three mechanisms
- Tissue disruption (brush): A brush component at the device tip physically disrupts the endometrial surface to loosen tissue, similar in concept to how a cervical brush works in a Pap smear.
- Suction (plunger-driven aspiration): A plunger inside the sheath generates suction that aspirates dislodged cells and tissue into the collection channel, preventing sample loss. This replaces the piston-retraction mechanism of the Pipelle with a more controlled aspiration system.
- Sponge absorption: A sponge tip at the device end absorbs fluid and cells from the uterine cavity. This captures material that would not be collected by suction alone, including cells suspended in uterine fluid rather than adherent to the wall.
The device features a slim-profile wand and an integrated cervical guard to prevent over-insertion. The company states the procedure is completed in approximately 20 seconds, requires no cervical dilation, no sedation, and no operating room.
What types of testing the sample supports
Because the device collects tissue through three complementary mechanisms, the resulting sample supports three categories of laboratory analysis from a single collection pass:
- Cytologic analysis: examination of individual cells and cell clusters under a microscope, comparable to cervical cytology in a Pap smear.
- Histopathologic analysis: examination of tissue architecture and cell morphology in the standard endometrial biopsy format, allowing diagnosis of endometrial hyperplasia, atypical hyperplasia, and carcinoma.
- Molecular analysis: biomarker testing including mismatch repair protein immunohistochemistry, Lynch syndrome screening, and other emerging molecular markers that increasingly inform endometrial cancer subtyping and treatment planning.
Artera notes that results are available within one to two days and that the test produces no inconclusive results based on insufficient tissue, which is a relevant distinction: one of the main failpoints of the current Pipelle is producing an insufficient sample that requires a return visit or D&C.
The Preclinical Evidence: What Testing Showed
The FDA clearance was supported by preclinical and design verification testing conducted by Medical Murray, a medical device manufacturer. Testing compared the Utepreva device against a commercially available endometrial sampler using a standardized model of simulated endometrial tissue under controlled conditions.
Under those conditions, the Utepreva device captured a greater volume of simulated tissue and demonstrated more uniform disruption across the sampling surface. The difference in tissue capture was statistically significant. The company has presented the device at the American College of Obstetricians and Gynecologists Annual Clinical and Scientific Meeting in May 2026.
| What the clearance pathway tells us about the evidence standard The FDA cleared Utepreva through the 510(k) pathway, which permits clearance of a medical device if it is substantially equivalent to an already legally marketed device. The Utepreva Endometrial Sampler is substantially equivalent to existing endometrial sampling devices, cleared for the same intended use, which is obtaining endometrial tissue samples for laboratory analysis. 510(k) clearance does not require the same level of clinical efficacy evidence as a PMA (premarket approval) or a drug NDA. The supporting data is preclinical bench testing, not randomized clinical trials in patients. This means the device’s performance in real clinical settings, across diverse patient populations and uterine anatomies, will need to be established through post-clearance use and publication. The absence of peer-reviewed clinical trial data at the time of clearance is a standard feature of most new medical device clearances, not a red flag specific to Utepreva. It is, however, a limitation worth naming clearly for anyone following this device’s evidence trajectory. |
What Patients Should Know: Who Needs Endometrial Evaluation and When
There is no routine screening test for average-risk women
Current guidelines from AAFP, ACOG, and the American Cancer Society do not recommend routine endometrial cancer screening in asymptomatic women at average risk. No Pap smear equivalent exists for the endometrium. This means that for most women, the pathway to early diagnosis runs through symptom recognition and timely evaluation, not through a scheduled test.
Report any postmenopausal bleeding promptly
Postmenopausal bleeding is the reason for approximately two-thirds of all gynecologic office visits in postmenopausal women, and it is the single most important early symptom of endometrial cancer. Any bleeding after 12 consecutive months without a period warrants a same-cycle evaluation rather than a wait-and-see approach. Even a single episode of light spotting should be discussed with a gynecologist.
Special situations that warrant proactive discussion
- Women with Lynch syndrome should discuss an individualized surveillance plan with their gynecologist. Annual endometrial sampling beginning at age 30 to 35 is considered for Lynch carriers in some guidelines, though the evidence base for specific protocols remains limited.
- Women taking tamoxifen should be counseled on endometrial cancer symptoms at each follow-up visit. Any abnormal uterine bleeding should trigger evaluation, even if the ovaries are still functioning.
- Women with obesity who are approaching or in the menopause transition have enough baseline risk that any menstrual irregularity outside a normal perimenopause pattern warrants discussion with a provider rather than dismissal.
When will Utepreva be available?
Utepreva LLC has announced the device will be available to healthcare providers beginning in October 2026. Patients will not purchase or use it directly. If your gynecologist performs endometrial sampling in the office, you can ask whether they will be adopting the new device. For now, the Pipelle and similar existing samplers remain the standard of care for in-office endometrial biopsy.
The bottom line
Endometrial cancer is the most common gynecologic cancer in the United States, and it is one where early detection reliably leads to good outcomes. The current diagnostic infrastructure relies on a device that has not been meaningfully updated in decades, and on patients and providers taking postmenopausal bleeding seriously at first presentation. The Utepreva Endometrial Sampler does not change who needs evaluation or when. What it offers, if its preclinical performance translates to clinical practice, is a more comprehensive tissue sample from a single office procedure. Real-world clinical data after the October 2026 launch will determine whether the promise holds. For patients and providers navigating this space today, the most useful resources remain ACOG’s clinical practice guidelines on endometrial cancer evaluation and the American Cancer Society’s endometrial cancer overview.
Sources
Utepreva press release (PR Newswire): Utepreva Introduces FDA 510(k)-Cleared Endometrial Sampler Designed to Support Early Detection of Endometrial Cancer. April 22, 2026.
Contemporary OB/GYN: Utepreva Launches FDA-Cleared Endometrial Sampler to Support Endometrial Cancer Detection. contemporaryobgyn.net. April 2026.
Clinical Lab Products: New Endometrial Sampling Device Receives FDA Clearance for Cancer Detection. clpmag.com. April 2026.
BioSpace: Utepreva Introduces FDA 510(k)-Cleared Endometrial Sampler. biospace.com. April 22, 2026.
FDA 510(k) clearance record: Utepreva Endometrial Sampler (UP01), K240595. FDA.gov.
AAFP Rapid Evidence Review: Endometrial Cancer. Am Fam Physician. 2025;111(6):526-531.
StatPearls (endometrial cancer): Endometrial Cancer. StatPearls. NCBI Bookshelf. Updated 2024.
StatPearls (postmenopausal bleeding): Postmenopausal Bleeding. StatPearls. NCBI Bookshelf. Updated January 2025.
Medscape: Endometrial Carcinoma: Background, Etiology, Epidemiology. emedicine.medscape.com.
ACS uterine cancer statistics: Key Statistics for Uterine Cancer. cancer.org.
| Disclaimer: Health Evidence Digest provides general information about medical devices and health research for educational purposes. This content is not a substitute for professional medical advice. The Utepreva Endometrial Sampler is a cleared medical device, not a diagnostic test or treatment. All decisions about endometrial evaluation and cancer screening should be made in consultation with a qualified gynecologist or healthcare provider. |