
| 📌 The essentials Drug: Loargys (pegzilarginase-nbln) — a PEGylated recombinant human arginase-1 enzyme. FDA approval type: Accelerated approval, February 23, 2026. Based on reduction of plasma arginine as a surrogate endpoint. Continued approval may depend on confirmatory trial results. Developer: Immedica Pharma (Stockholm/Chicago). Indication: Treatment of hyperargininemia in adult and pediatric patients 2 years of age and older with Arginase 1 Deficiency (ARG1-D), in conjunction with dietary protein restriction. What makes it first-in-class: First and only FDA-approved therapy proven to lower arginine levels in ARG1-D. Prior management was symptomatic only — dietary restriction and ammonia scavengers. Neither addressed endogenous arginine production. Key trial results (PEACE, n=32): 90.5% of pegzilarginase patients normalized plasma arginine at 24 weeks vs 0% on placebo. Mean plasma arginine reduced from 354 to 86 μmol/L (72% reduction, p<0.0001). Administration: Intravenous infusion once weekly, administered in a healthcare setting. Subcutaneous administration was offered in the long-term extension. Critical warning: Boxed warning for life-threatening hypersensitivity reactions including anaphylaxis. Requires administration under direct healthcare supervision with emergency support available. Availability: Estimated available in the U.S. from April 2026. Patient support: There for Rare program (Immedica). |
The parents of a child with Arginase 1 Deficiency learn quickly that the disease does not announce itself with a dramatic crisis. There is no sudden collapse. There is no dramatic fever. Instead, something subtler and harder to name: the child who was walking begins to walk differently. The legs become stiffer. The gait changes. Developmental milestones come later than expected, or not at all. The child who could run begins to struggle with stairs.
Many of these families spent years before diagnosis being told it was cerebral palsy, or hereditary spastic paraplegia, or developmental delay without a clear cause. The correct diagnosis, when it finally came, offered a name for what was happening but not a way to stop it. Management meant strict dietary protein restriction, which is genuinely difficult to maintain in a young child, and ammonia scavenger drugs that addressed one downstream consequence of the disease without touching the core problem: too much arginine in the blood, accumulating every day, slowly damaging the nervous system.
On February 23, 2026, the FDA granted accelerated approval to Loargys (pegzilarginase-nbln) for the treatment of hyperargininemia in patients aged 2 and older with Arginase 1 Deficiency. It is the first drug approved that directly addresses the underlying biochemical defect — replacing the missing enzyme, lowering arginine, and in long-term studies, improving the very spasticity that defines this disease.
What Is Arginase 1 Deficiency and Why Does Arginine Damage the Nervous System?
The urea cycle is the biochemical pathway the liver uses to convert ammonia, a toxic byproduct of protein metabolism, into urea for excretion. The cycle involves eight enzymes working in sequence. Arginase 1 is the final enzyme in the cycle: it cleaves the amino acid arginine into urea and ornithine, the last step before ammonia-derived waste leaves the body.
In ARG1-D, both copies of the ARG1 gene are mutated (autosomal recessive inheritance), and arginase-1 enzyme activity is severely reduced or absent. Arginine, which cannot be cleared, accumulates persistently in the blood and cerebrospinal fluid. Normal plasma arginine is below 100 micromolar (μmol/L). Patients with ARG1-D typically present with levels of 300 to 500 μmol/L. It is this chronic arginine elevation, and the accumulation of arginine’s toxic metabolites including guanidino compounds, that causes progressive neurological damage.
Unlike most other urea cycle disorders, ARG1-D is not primarily characterized by hyperammonemia crises. Ammonia levels are often near-normal, because the blocked urea cycle step is at the end of the pathway. This is part of why the disease is different from other urea cycle disorders and why managing ammonia does not meaningfully control ARG1-D.
| How ARG1-D typically presents and why it is so often misdiagnosed Clinical manifestations in ARG1-D are typically absent in the newborn period and early infancy, despite the metabolic defect being present from birth. Symptoms usually first appear between ages 1 and 3 years. The most common initial presentation is spasticity of the lower limbs, followed by developmental delay and loss of previously acquired motor milestones. Because spasticity and gait abnormalities in young children are common features of cerebral palsy and hereditary spastic paraplegia, ARG1-D is frequently misdiagnosed as one of these conditions. A systematic review found that diagnostic delays are common, driven by limited disease awareness, the absence of hyperammonemic crises (which would more immediately suggest a urea cycle disorder), and inconsistent availability of newborn screening programs that could detect elevated arginine at birth. ARG1-D is listed on the Recommended Uniform Screening Panel (RUSP) for U.S. newborn screening, but inclusion varies by state, and the sensitivity of screening for ARG1-D using standard amino acid profiling has been debated. For families who receive a diagnosis late, the neurological damage that has already accumulated during the years of missed diagnosis is not fully reversible. This is the clinical argument for early diagnosis and early treatment. |
How Loargys Works: Replacing the Enzyme, Not Just Managing Its Absence
Pegzilarginase is a recombinant human arginase-1 enzyme produced using genetic engineering, then modified by attaching polyethylene glycol (PEG) chains to its surface. This PEGylation serves two purposes: it extends the enzyme’s half-life in the bloodstream, making once-weekly dosing sufficient, and it reduces immunogenicity, lowering the risk that the immune system will produce antibodies that neutralize the enzyme.
When pegzilarginase is administered intravenously, it acts as an exogenous source of the arginase-1 activity the patient’s own cells cannot provide. It circulates in the blood and catalyzes the conversion of arginine to ornithine and urea, just as endogenous arginase-1 would in a person without the deficiency. This directly lowers plasma arginine levels toward the normal range.
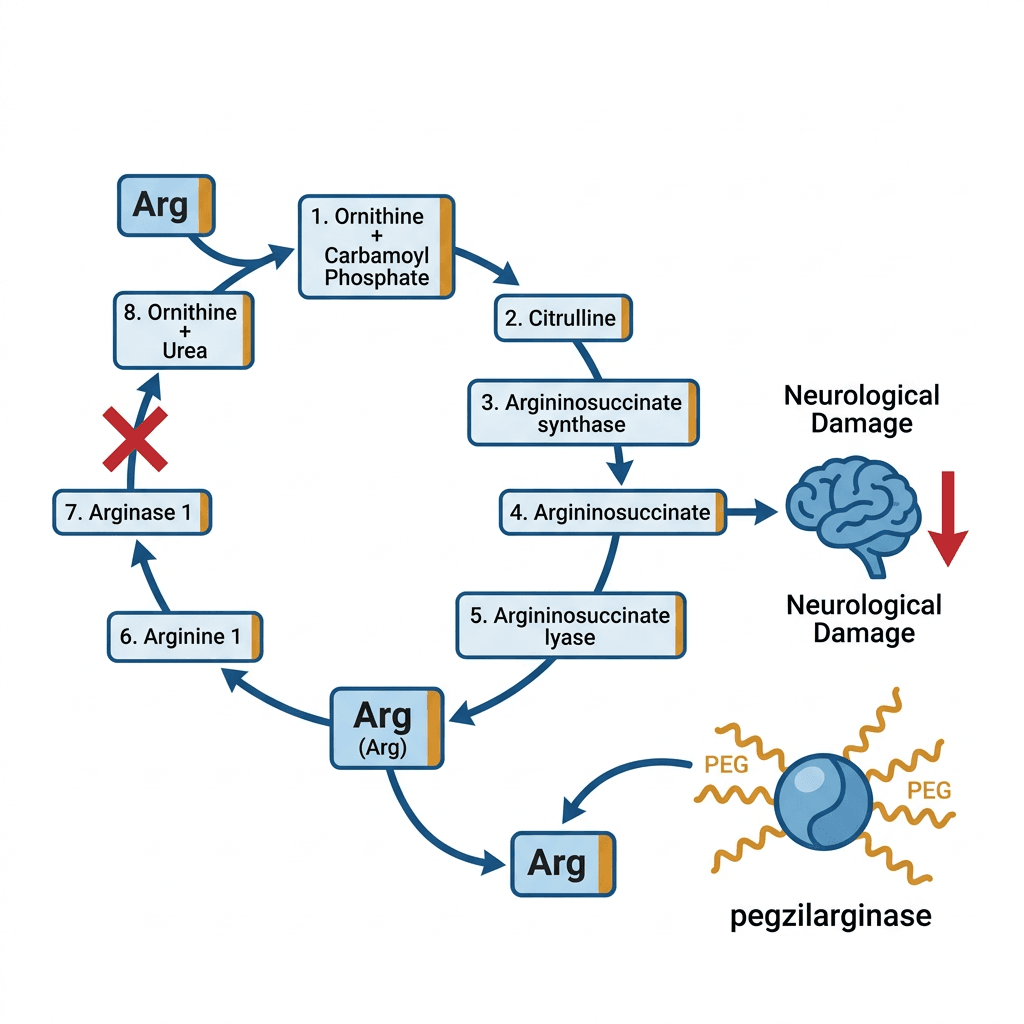
This mechanism addresses the fundamental limitation of dietary protein restriction: even with strict dietary control, the body continues to produce arginine endogenously through the first part of the urea cycle. Restricting dietary protein reduces the amount of arginine coming in from food, but it cannot stop the liver from producing arginine from within. Pegzilarginase clears both sources.
The PEACE Trial: What the Clinical Data Shows
Trial design
The PEACE trial (NCT03921541) was a Phase 3, randomized, double-blind, placebo-controlled study conducted at 19 sites in 7 countries (United States, Canada, United Kingdom, Austria, France, Germany, and Italy). It enrolled 32 patients aged 2 years and older with genetically confirmed ARG1-D. Patients were randomized 2:1 to receive IV pegzilarginase (n=21) or matched placebo (n=11) once weekly for 24 weeks. Patients continued their existing individualized disease management (dietary restriction, ammonia scavengers) throughout.
A sample size of 32 in a randomized controlled trial is small by conventional standards. It is large for a disease with a median prevalence of 1 in 1,000,000. The PEACE trial enrolled what was, at the time, the largest prospectively studied ARG1-D cohort ever assembled, and it is the first randomized, blinded, placebo-controlled clinical trial ever conducted in this disease.
Primary endpoint: plasma arginine reduction
| Measure | Pegzilarginase | Placebo |
| Plasma arginine at baseline | 354.0 μmol/L (geometric mean) | 464.7 μmol/L |
| Plasma arginine at week 24 | 86.4 μmol/L | 426.6 μmol/L |
| Mean percent reduction | 72% (p<0.0001) | No meaningful change |
| Mean absolute reduction | −312 μmol/L (95% CI −384 to −239) | — |
| Patients reaching target (<200 μmol/L) | 90.5% | 0% |
| Patients reaching normal levels (<100 μmol/L) | 90.5% | 0% |
Source: PEACE trial, eClinicalMedicine (Lancet). 2024;68:102400. doi:10.1016/j.eclinm.2023.102400. NCT03921541.
The 90.5% versus 0% normalization rate is the most arresting number in the entire dataset. No patient on placebo brought their plasma arginine into the normal range; nearly all patients on pegzilarginase did. The clinical trial design, in which dietary management continued in both arms, means this difference is attributable specifically to the enzyme replacement therapy, not to dietary changes.
Mobility outcomes: secondary endpoints and long-term extension data
The primary endpoint of the PEACE trial was plasma arginine reduction, the surrogate measure on which accelerated approval was granted. Secondary endpoints examined functional mobility using validated instruments: the Gross Motor Function Measure (GMFM-D and GMFM-E subscales), timed walk tests, and the 6-minute walk test. These endpoints showed a positive trend in the pegzilarginase arm during the 24-week trial period.
The more clinically compelling functional data comes from the long-term extension (LTE) studies, which followed patients receiving pegzilarginase for up to 5 years. Published in 2025, these combined results from Study 102A (n=14, up to 5 years) and the PEACE LTE (n=31, up to 3 years) showed:
- Spasticity improved in 84% of patients, as measured by the Modified Ashworth Scale (MAS).
- 12 patients achieved MAS 0, meaning no detectable spasticity at all.
- Six-minute walk test (6MWT) distances improved across the cohort, reflecting real-world functional mobility gains.
- Gross Motor Function Measure scores improved in the D and E subscales (standing and walking/running/jumping), which are the most clinically relevant for patients with ARG1-D.
- Plasma arginine remained suppressed at normal or near-normal levels throughout the extension period.
- An Italian real-world case series also documented that when treatment was interrupted for 13 months after the trial concluded, arginine levels returned and spasticity worsened; when treatment was restarted, benefits resumed. This discontinuation-rechallenge observation is the strongest indirect evidence that the biochemical normalization drives the functional improvement.
| Why this is accelerated approval and what that means The FDA approved Loargys under the accelerated approval pathway, which allows earlier access to drugs for serious conditions based on a surrogate endpoint reasonably likely to predict clinical benefit. For Loargys, the surrogate is plasma arginine reduction. The rationale is well-established in the ARG1-D literature: persistent arginine elevation is the proximal driver of neurological damage in this disease. Normalizing arginine is mechanistically sound as a predictor of clinical benefit. Continued approval may depend on confirmatory trial results demonstrating actual clinical benefit such as improved functional outcomes. The long-term extension data described above, while not the confirmatory trial, provides the strongest available evidence that arginine normalization does translate into meaningful spasticity reduction and mobility improvement. For families: accelerated approval means the FDA has determined the drug works on a meaningful surrogate and that the benefit outweighs the risk given the severity of the disease. It does not mean clinical benefit is fully confirmed. The full evidence picture will emerge from the confirmatory studies. |
Safety: The Anaphylaxis Warning That Requires In-Clinic Administration
Boxed warning: hypersensitivity reactions including anaphylaxis
Loargys carries a boxed warning for life-threatening hypersensitivity reactions, including anaphylaxis. This risk is the primary reason the drug must be administered under direct supervision in a healthcare setting with emergency support immediately available. Reactions can occur during or after infusion. For this reason, Loargys is not a self-administered home therapy — every dose requires a scheduled clinic or infusion center visit.
Signs of a hypersensitivity reaction that require immediate intervention include hives, difficulty breathing, swelling of the face or throat, rapid or irregular heartbeat, dizziness, and loss of consciousness. Patients and families should be counseled on these symptoms before each infusion.
Other adverse reactions
The most common adverse reactions in the PEACE trial were vomiting, pyrexia (fever), infusion-associated reactions, and constipation. These are manageable in most cases. The real-world Italian case series, following three pediatric patients over years of treatment, did not report serious safety events apart from infusion reactions. The prescribing information also includes a precaution for embryo-fetal toxicity, relevant for patients of reproductive age.
Practical Details: Dosing, Administration, and Patient Support
| Feature | Details |
| Route | Intravenous infusion (30 minutes) |
| Frequency | Once weekly |
| Setting | Healthcare setting with direct supervision; emergency support required |
| Available formulations | Single-dose vials: 2 mg/0.4 mL and 5 mg/1 mL |
| Subcutaneous option | Available in long-term extension phase; ask treating metabolic specialist about current access |
| Used alongside | Dietary protein restriction (required); ammonia scavengers if applicable |
| Availability | Estimated available in U.S. from April 2026 |
| Patient support | There for Rare program (Immedica): financial assistance and nonmedical education support |
| Contraindications | Known hypersensitivity to pegzilarginase or excipients |
| Pediatric use | Approved from age 2 years; most clinical trial participants were pediatric or young adults |
What This Means for Families Affected by ARG1-D
For a disease affecting roughly 1 in 1,000,000 people, ARG1-D has generated a disproportionate amount of research attention over the past decade, driven substantially by patient advocacy. The Arginase 1 Deficiency Foundation, established by families directly affected by the disease, has been one of the forces accelerating clinical development and raising the standard of care expectations for this patient community.
The clinical significance of this approval should be understood in its proper context. Most patients diagnosed with ARG1-D today are children or young adults who have already accumulated some neurological damage before treatment can begin, because diagnosis often comes late. Loargys does not reverse established neurological damage. What the long-term extension data shows is that it can halt or reduce ongoing spasticity progression and improve functional mobility in patients who begin treatment. The most meaningful benefit likely comes with early treatment, before accumulated damage is severe.
For newly diagnosed patients identified through newborn screening or early clinical presentation, Loargys represents the possibility of intervening before significant neurological damage accumulates. For older patients who have been managing on dietary restriction and ammonia scavengers alone, it represents the first drug that can actually bring arginine to normal levels and the only approved therapy with evidence of functional benefit.
The weekly infusion requirement is a real burden for families, many of whom already manage complex dietary protocols and multiple specialist appointments. The availability of subcutaneous administration in the extension period, if it eventually becomes part of the approved label, would substantially reduce that burden. This is worth monitoring as post-marketing data accumulates.
Resources for families and clinicians
For families navigating an ARG1-D diagnosis, the Arginase 1 Deficiency Foundation (a1d.org) provides patient and family support resources, clinical trial information, and a community of families with direct experience of this disease. The National Urea Cycle Disorders Foundation (nucdf.org) covers the full spectrum of urea cycle disorders and maintains a specialist referral directory. For Loargys access and patient support, the There for Rare program is available through immedicaus.com.
Sources
FDA accelerated approval announcement: FDA grants accelerated approval to pegzilarginase-nbln (Loargys) for treatment of hyperargininemia in patients with ARG1-D. FDA.gov. February 23, 2026.
Immedica press release: U.S. FDA has granted accelerated approval of Loargys (pegzilarginase-nbln) for treatment of hyperargininemia in patients 2 years and older with Arginase 1 Deficiency. Immedica Pharma. February 23, 2026.
Endocrinology Advisor: FDA Grants Accelerated Approval to Loargys for Arginase 1 Deficiency. endocrinologyadvisor.com. February 25, 2026.
Drugs.com history: Loargys (pegzilarginase-nbln) FDA Approval History. drugs.com.
PEACE trial primary publication (eClinicalMedicine): Efficacy and safety of pegzilarginase in arginase 1 deficiency (PEACE): a phase 3, randomized, double-blind, placebo-controlled, multi-centre trial. eClinicalMedicine (The Lancet). 2024;68:102400. doi:10.1016/j.eclinm.2023.102400. PMC10825663.
Long-term extension study: Long-Term Efficacy and Tolerability of Pegzilarginase in Arginase 1 Deficiency: Results of Two International Multicentre Open-Label Extension Studies. PubMed PMID 40714964.
Italian real-world case series (MDPI): Pegzilarginase in Arginase 1 Deficiency: Clinical and Biochemical Effects of Treatment Initiation, Discontinuation and Re-Initiation. MDPI Children. 2026;13(5):610.
ARG1-D systematic review (prevalence/diagnosis): Epidemiology, methods of diagnosis, and clinical management of patients with ARG1-D: A systematic review. PubMed PMID 36049366.
Natural history systematic review: Natural history of arginase 1 deficiency and the unmet needs of patients: A systematic review of case reports. JIMD Reports. PMC9259395.
Misdiagnosis/clinical review (PMC): Arginase 1 deficiency: a treatable form of spastic paraplegia. PMC12394256.
Trial registration: PEACE: A Phase 3 Study of Pegzilarginase in Patients With Arginase 1 Deficiency. NCT03921541. clinicaltrials.gov.
Patient resources: Arginase 1 Deficiency Foundation: a1d.org; National Urea Cycle Disorders Foundation: nucdf.org; Immedica patient support: immedicaus.com
| Disclaimer: Health Evidence Digest provides general information about FDA approvals and health research for educational purposes. This content is not a substitute for professional medical advice. Loargys is approved under the accelerated approval pathway, and continued approval may depend on confirmatory trial results. Treatment decisions for ARG1-D should be made in consultation with a metabolic specialist or biochemical geneticist experienced in urea cycle disorders. |
Leave a Reply