| 📌 The essentials On April 30, 2026, the FDA’s Oncologic Drugs Advisory Committee (ODAC) voted 6 to 3 against the clinical benefit of switching to camizestrant (AstraZeneca) in patients with HR-positive, HER2-negative metastatic breast cancer upon detection of an emerging ESR1 mutation during first-line therapy, before radiographic disease progression. The vote was based on data from the SERENA-6 Phase 3 trial. One day later, on May 1, 2026, the FDA approved vepdegestrant (Veppanu, Arvinas/Pfizer) for ER-positive, HER2-negative, ESR1-mutated advanced breast cancer after prior endocrine therapy. Both decisions involve ESR1 mutations in the same general patient population. They reached opposite conclusions. This post explains why, and what the difference reveals about how the FDA evaluates evidence in precision oncology. |
|---|
Within 24 hours in late April and early May 2026, the FDA’s approach to ESR1-guided breast cancer treatment produced two very different outcomes. On the same day that an advisory panel voted against approving camizestrant for a ctDNA-guided treatment switch before disease progression, vepdegestrant was on its way to full FDA approval for the same patient population at a later stage of treatment. Understanding why these two decisions went in opposite directions requires understanding exactly what each drug was asking the FDA to accept.
What ESR1 Mutations Are and Why They Matter
ESR1 mutations occur in the gene that encodes the estrogen receptor. In patients with hormone receptor-positive, HER2-negative breast cancer, the estrogen receptor is the primary driver of tumor growth, which is why endocrine therapies that block or degrade it form the backbone of treatment.
The problem is that treatment pressure on the estrogen receptor eventually selects for mutations that allow it to remain active even in the absence of estrogen. These ESR1 mutations are acquired, meaning they typically arise during treatment rather than being present at diagnosis. They are detected in approximately 40 to 50% of patients who progress on first-line endocrine therapy plus a CDK4/6 inhibitor. When they emerge, they signal developing resistance and predict poor outcomes on continued aromatase inhibitor-based therapy.
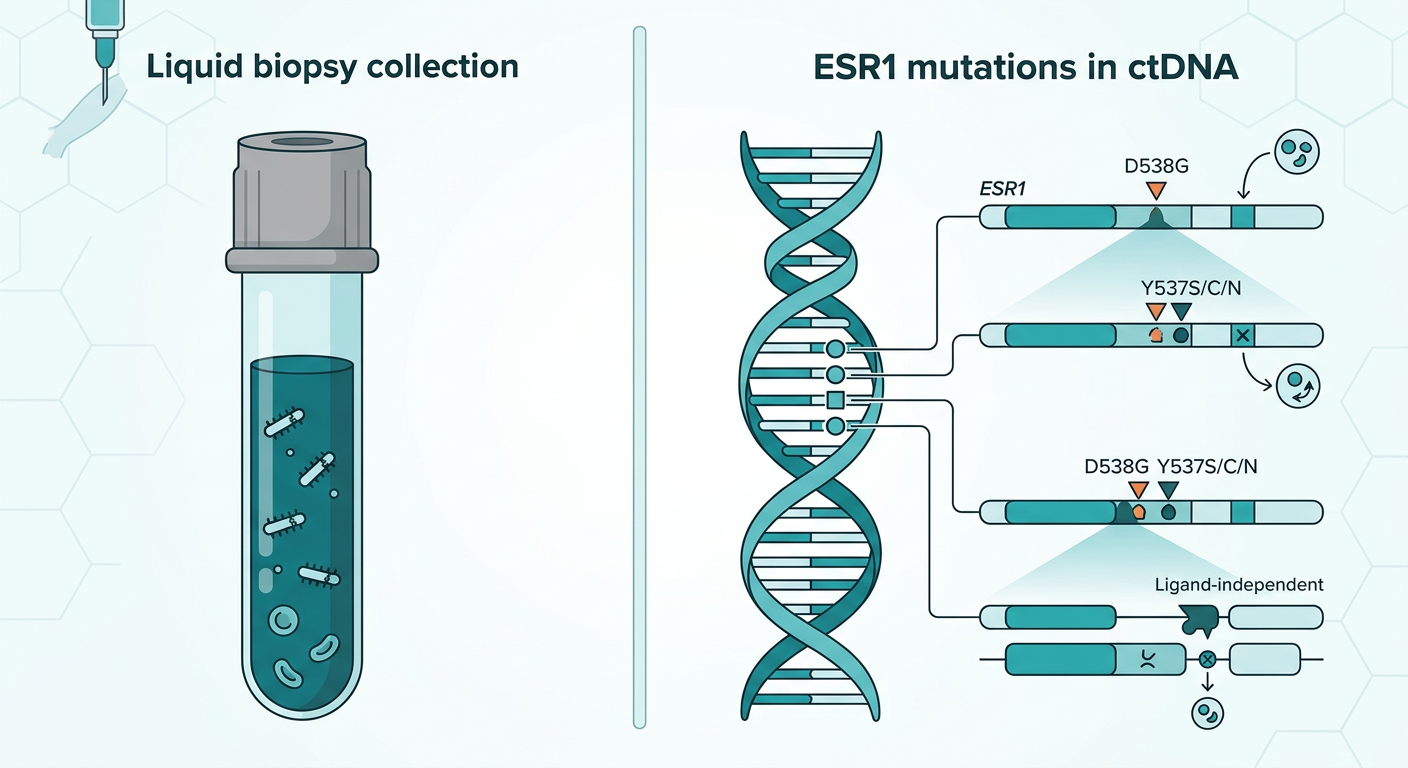
Liquid biopsy technology, specifically circulating tumor DNA (ctDNA) testing, can now detect these mutations from a blood draw, often before the tumor shows measurable growth on a scan. That capability is central to both of the regulatory stories described in this post, but in two very different ways.
Vepdegestrant: The Approval That Happened
On May 1, 2026, the FDA approved vepdegestrant (Veppanu) for adults with ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer who had disease progression following at least one line of endocrine therapy. The approval arrived more than a month ahead of the June 5 PDUFA date, a signal that the FDA’s review was straightforward.
The approval simultaneously authorized the Guardant360 CDx liquid biopsy as a companion diagnostic to identify patients with ESR1 mutations who are eligible for treatment.
What the VERITAC-2 Trial Showed
The approval was based on data from VERITAC-2 (NCT05654623), a global, randomized, open-label Phase 3 trial that enrolled 624 patients at 213 sites across 25 countries. Patients were required to have disease progression on one to two lines of endocrine therapy, including one line with a CDK4/6 inhibitor. They were randomized 1:1 to receive either vepdegestrant orally once daily or fulvestrant intramuscularly.
In the 270-patient ESR1-mutated subgroup that drove the approval, vepdegestrant reduced the risk of disease progression or death by 43% compared to fulvestrant, with a median progression-free survival of 5.0 months versus 2.1 months (hazard ratio 0.57; 95% CI 0.42 to 0.77; p=0.0001). Across the overall trial population, regardless of ESR1 status, the PFS benefit did not reach statistical significance (hazard ratio 0.83; p=0.07), which reinforces the importance of ESR1 mutation testing before treatment selection and underscores that this approval is strictly for the ESR1-mutated population. Overall survival data are still immature, with only 16% of deaths having occurred at the time of the PFS analysis.
The VERITAC-2 results were presented at the 2025 ASCO Annual Meeting and simultaneously published in The New England Journal of Medicine.
What Makes Vepdegestrant Mechanistically Different
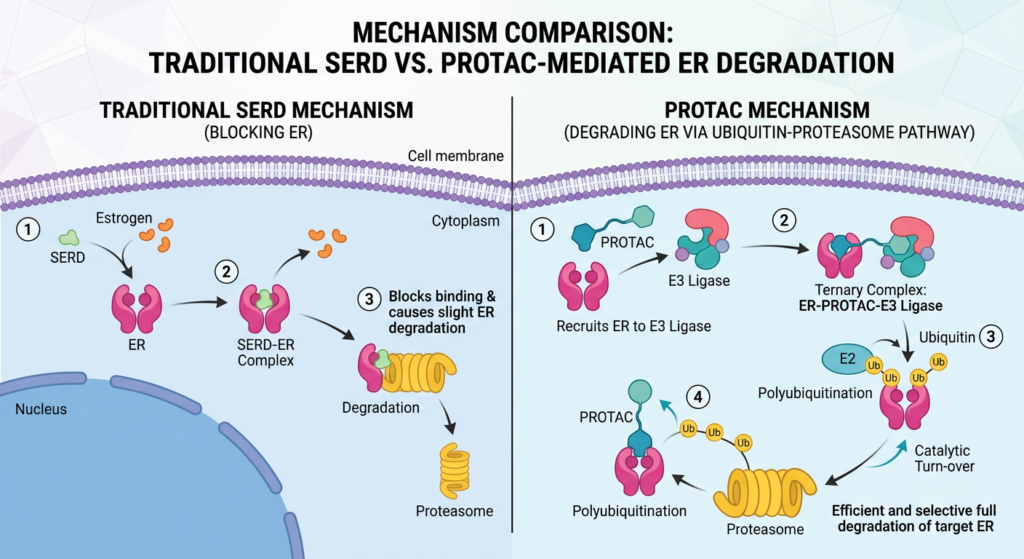
Vepdegestrant is more than a new drug in an existing class. It is the first PROTAC (proteolysis-targeting chimera) to receive FDA approval for any indication, making this a landmark regulatory event beyond its breast cancer-specific context.
Traditional SERDs (selective estrogen receptor degraders) like fulvestrant and elacestrant work by binding the estrogen receptor and triggering its degradation. Vepdegestrant takes a different approach: it is a bifunctional molecule that simultaneously recruits the estrogen receptor on one end and a cellular protein-degradation machinery component called an E3 ubiquitin ligase on the other. By bringing these two proteins into proximity, it directs the cell’s own waste-disposal system to destroy the estrogen receptor completely rather than simply blocking it.
This catalytic mechanism means one molecule of vepdegestrant can degrade multiple copies of the estrogen receptor and is then recycled to degrade more. It eliminates the receptor rather than occupying it, which is mechanistically important when dealing with ESR1 mutations that cause the receptor to remain active even when blocked.
We covered the full PROTAC mechanism and the VERITAC-2 trial data in detail here. The May 1 FDA approval means that post is now confirmed, and the drug is commercially available.
What Patients Should Know About Vepdegestrant
Vepdegestrant (Veppanu) is an oral once-daily tablet taken with food at a dose of 200 mg. It is indicated for patients who have already progressed on at least one line of endocrine therapy, including a CDK4/6 inhibitor. ESR1 mutation testing with an FDA-authorized ctDNA assay such as Guardant360 CDx is required before starting treatment.
The FDA label includes warnings about QTc interval prolongation (a heart rhythm consideration that requires monitoring) and embryo-fetal toxicity. The most common adverse effects across the trial were musculoskeletal pain, nausea, fatigue, hot flashes, and headache. Patients should discuss the full safety profile with their oncologist.
Camizestrant: The Vote Against
The ODAC vote on April 30, 2026, addressed a very different question. AstraZeneca was not asking the FDA to approve camizestrant for patients who had already progressed. It was asking whether camizestrant should be approved for patients who had developed an ESR1 mutation in a ctDNA blood test but had not yet shown radiographic evidence of disease progression on their current treatment.
This is a fundamentally different clinical scenario, and the distinction is the reason the vote went against approval.
The SERENA-6 Trial Design
SERENA-6 (NCT04964934) enrolled patients with HR-positive, HER2-negative advanced breast cancer who were stable on first-line aromatase inhibitor plus CDK4/6 inhibitor therapy for at least six months. Every two to three months, patients had ctDNA testing using the Guardant360 CDx assay. When an ESR1 mutation was detected in the blood, patients who had no evidence of disease progression on imaging were randomized to either continue their existing therapy or switch to camizestrant 75 mg plus their CDK4/6 inhibitor.
The PFS results were numerically compelling. The median PFS was 16.0 months in the camizestrant arm versus 9.2 months in the continued-aromatase-inhibitor arm (hazard ratio 0.44; p less than 0.00001). By the conventional statistical measures, this looks like a large effect. The ODAC voted 6 to 3 against it anyway. Why?
Why ODAC Said No
The committee’s concerns centered on three interconnected problems with interpreting the trial’s results as evidence of clinically meaningful benefit.
The PFS time zero problem. In SERENA-6, progression-free survival was measured from the time of randomization, which occurred at ESR1 mutation detection rather than at the start of treatment. Patients in the control arm who were still on therapy at randomization were inevitably closer to their next progression event than patients who had just started a new drug. This creates a structural asymmetry in how PFS is measured across the two arms that is not a drug effect. FDA reviewers flagged this as a nonstandard PFS time zero that complicates interpretation.
PFS2 is confounded by the protocol design. PFS2 (time to progression on the next line of therapy) is sometimes used as a supporting endpoint to demonstrate that a PFS benefit translates downstream. In SERENA-6, patients in the control arm were switched to camizestrant upon progression, as specified in the protocol. This protocol-mandated switch means PFS2 cannot serve as an independent confirmation of benefit, because both arms ultimately received the same drug.
Overall survival is immature and uncertain. OS data at the time of the ODAC meeting were too early to be interpretable. Committee members noted that without a mature OS signal, and with the PFS data carrying the methodological concerns described above, there was insufficient evidence that the ctDNA-guided switch before progression meaningfully improved patient outcomes compared to simply switching at the time of standard radiographic progression.
One additional safety note that ODAC discussed: there is a signal of potential cardiac toxicity when camizestrant is combined with ribociclib, one of the CDK4/6 inhibitors used in the trial. This was not a primary reason for the negative vote, but it added to the committee’s caution.
The Conceptual Question at the Heart of Both Decisions
Camizestrant and vepdegestrant both target ESR1 mutations. Both are oral SERDs (camizestrant) or SERD-class agents (vepdegestrant). But they were asking the FDA to accept fundamentally different propositions.
Vepdegestrant asked: does this drug help patients after they have already progressed on prior therapy? This is a well-established clinical endpoint with clear time zero, an appropriate comparator (fulvestrant, which is the current standard), and a patient population that has a demonstrated unmet need. The answer was yes.
Camizestrant asked: should treatment be switched based on a molecular signal in the blood, before the patient shows any clinical or radiographic signs of progression? This is a newer paradigm in precision oncology called ctDNA-guided adaptive therapy, and the SERENA-6 trial was the first global registrational trial to test it. The FDA’s position, reflected in the ODAC vote, was that SERENA-6 did not adequately answer this question.
The comparison matters because it illustrates a distinction that runs through many recent oncology regulatory debates. A large PFS hazard ratio is not by itself sufficient evidence of clinical benefit if the design creates ambiguity about what is actually being measured. ODAC members were not questioning whether camizestrant is an active drug. Several acknowledged that the drug likely has meaningful anti-tumor activity. What they were questioning is whether the SERENA-6 trial design adequately demonstrated that switching before progression is better for patients than switching at progression, which is the standard approach.
What Happens to Camizestrant Next
An ODAC vote against clinical benefit does not automatically result in a formal FDA rejection, but it is a strong signal. Given that the FDA’s own reviewers raised similar concerns before the advisory committee meeting, approval of the specific indication tested in SERENA-6 is unlikely without additional data.
AstraZeneca has separate ongoing trials for camizestrant in different settings. The SERENA-4 trial is evaluating camizestrant in the first-line setting for HR-positive, HER2-negative advanced breast cancer, which is a different clinical question and regulatory submission. A negative ODAC vote on one trial in one specific indication does not preclude a different regulatory outcome for the same drug in a different setting.
What ctDNA-Guided Treatment Really Means for the Field
SERENA-6 was the first randomized registrational trial to test the concept of using a liquid biopsy molecular signal to guide a treatment switch before clinical progression. The ODAC vote does not close the door on this concept. It identifies what the evidence will need to look like for regulators to accept it.
The fundamental question is whether catching and responding to molecular resistance earlier than radiographic progression meaningfully changes patient outcomes. The biological rationale is plausible: treating a smaller, less heterogeneous tumor burden before the clone driving resistance has fully taken over should theoretically be advantageous. But biological plausibility and clinical proof are different things. The trial design challenges in SERENA-6 made it difficult for the committee to separate the drug’s effect from the structural features of the ctDNA-guided randomization approach.
Future trials in this space will likely need to address the PFS time zero issue directly, use OS or PFS2 endpoints that are not confounded by protocol-mandated switches, and potentially show head-to-head evidence that pre-progression switching outperforms standard progression-triggered switching. That is a harder evidentiary bar, but it is a consistent one. The FDA has applied similar rigor to other adaptive precision oncology designs.
For patients with HR-positive metastatic breast cancer and their oncologists, the practical takeaway is that ctDNA testing for ESR1 mutations is increasingly central to treatment decisions. The Guardant360 CDx approval as a companion diagnostic for vepdegestrant means ESR1 liquid biopsy testing is now both clinically actionable and reimbursement-supported in the context of that approved indication. Whether it will also be used to guide pre-progression switches remains an open question pending further evidence.
What to Ask Your Oncologist
If you have HR-positive, HER2-negative advanced or metastatic breast cancer and have progressed on prior endocrine therapy including a CDK4/6 inhibitor, ask whether ESR1 mutation testing has been done on a recent blood sample. If an ESR1 mutation is present, vepdegestrant (Veppanu) is now an approved option and should be part of the treatment discussion.
If you are currently stable on first-line aromatase inhibitor plus CDK4/6 inhibitor therapy, the ctDNA-guided pre-progression switch with camizestrant is not currently an approved approach based on the April 30 ODAC vote. Routine ESR1 monitoring during first-line therapy is likely to be discussed by your oncologist as evidence continues to develop, but it should not change your current treatment plan without a direct conversation with your care team.
For information on clinical trials evaluating newer approaches to ESR1-mutated breast cancer, ClinicalTrials.gov is the primary reference.
Sources
FDA approval of vepdegestrant: FDA approves vepdegestrant for ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer. FDA.gov. May 1, 2026.
VERITAC-2 trial registration: NCT05654623. ClinicalTrials.gov.
Arvinas FDA approval announcement: Arvinas Announces FDA Approval of VEPPANU (vepdegestrant). GlobeNewswire. May 1, 2026.
Targeted Oncology vepdegestrant approval: FDA Approves Vepdegestrant for ESR1-Mutated ER+/HER2- Advanced Breast Cancer. Targeted Oncology. May 2026.
OncLive ODAC vote coverage: FDA ODAC Votes Against Clinical Benefit of Switching to Camizestrant in HR+ Breast Cancer After ESR1 Mutation Detection. OncLive. April 30, 2026.
CancerNetwork ODAC coverage: FDA ODAC Votes No to Camizestrant for HR+/HER2- ESR1 Advanced Breast Cancer. CancerNetwork. April 2026.
Targeted Oncology ODAC coverage: FDA’s ODAC Votes Against Camizestrant in Advanced Breast Cancer. Targeted Oncology. April 2026.
AstraZeneca press release on ODAC vote: Update on FDA Advisory Committee vote on camizestrant. AstraZeneca. April 30, 2026.
OncLive April breast cancer flashback: FDA Flashback: Breast Cancer Decisions and News From April 2026. OncLive. 2026.
SERENA-6 trial registration: NCT04964934. ClinicalTrials.gov.
ESR1 mutation background: ESR1 mutations in breast cancer. StatPearls. NCBI.
AACR Q1 2026 Approvals: FDA Approvals in Oncology: January-March 2026. AACR Cancer Research Catalyst. April 2026.
| Disclaimer: Health Evidence Digest provides general information about FDA regulatory processes, clinical trial results, and oncology research for educational purposes only. Nothing on this site constitutes medical advice, diagnosis, or treatment. Vepdegestrant (Veppanu) is FDA-approved and commercially available; camizestrant is not approved in the indication discussed in this post. Treatment decisions for advanced breast cancer should be made in close consultation with a qualified oncologist who can evaluate your individual diagnosis, mutation status, and treatment history. |
|---|