| ✅ Updated May 19, 2026: FDA Approval Confirmed On May 15, 2026, the FDA approved two new indications for Enhertu (fam-trastuzumab deruxtecan-nxki, T-DXd) in adults with HER2-positive early-stage breast cancer. This post has been updated throughout to reflect both approvals. For related coverage of Enhertu’s approved indications in metastatic HER2-positive and HER2-low breast cancer, and how this new approval extends Enhertu into the curative-intent setting, see our companion post on Dato-DXd in triple-negative breast cancer for context on the broader ADC landscape. |
|---|
| 📌 The essentials: Two approvals, two different clinical situations Indication 1: Neoadjuvant (before surgery) FDA approved Enhertu followed by THP (taxane, trastuzumab, pertuzumab) for adults with HER2-positive (IHC 3+ or ISH+) stage II or III breast cancer before surgery. Dose: 5.4 mg/kg IV every 3 weeks for 4 cycles, then THP for 4 cycles, then surgery. Clinical basis: DESTINY-Breast11 (NCT05113251): pCR rate 67.3% with T-DXd plus THP versus 56.3% with standard anthracycline-based ddAC-THP (absolute improvement +11.2%; p=0.003). Indication 2: Adjuvant (after surgery, for residual disease) FDA approved Enhertu for adults with HER2-positive (IHC 3+ or ISH+) breast cancer who have residual invasive disease after neoadjuvant HER2-targeted treatment. Dose: 5.4 mg/kg IV every 3 weeks for a maximum of 14 cycles. Clinical basis: DESTINY-Breast05 (NCT04622319): T-DXd reduced the risk of invasive disease recurrence or death by 53% versus T-DM1 (Kadcyla) (HR 0.47; 95% CI 0.34 to 0.66; p less than 0.0001). Three-year invasive disease-free survival (iDFS): 92.4% versus 83.7%. Key safety note: the label carries a boxed warning for interstitial lung disease (ILD) and pneumonitis. ILD rate was 4.4% in DESTINY-Breast11 and approximately 10% in DESTINY-Breast05. Additional warnings: neutropenia and left ventricular dysfunction. |
|---|
When a woman is diagnosed with HER2-positive breast cancer, the weeks between diagnosis and surgery are not a waiting period. They are a treatment window, one that oncologists have spent decades trying to use more aggressively and more effectively. The drugs given before surgery, in the neoadjuvant setting, have the opportunity to shrink the tumor, treat any cancer that may have spread to lymph nodes or beyond, and ideally produce an outcome that changes what surgery looks like and what long-term prognosis looks like.
That neoadjuvant window has been anchored to anthracycline-containing chemotherapy regimens for more than a decade. Anthracyclines work, but they carry a burden: cardiac toxicity, hematologic toxicity, significant treatment interruptions. The question oncologists have been asking is whether something better is now available.
On May 18, 2026, the FDA is expected to rule on exactly that question. The drug at the center of the decision is Enhertu (trastuzumab deruxtecan, T-DXd), an antibody-drug conjugate that has already redefined outcomes in metastatic breast cancer. The clinical trial behind the application, DESTINY-Breast11, is the first positive global registrational trial for a new neoadjuvant agent in HER2-positive early breast cancer in over a decade. The data makes a compelling case. Understanding what it actually shows, and what it doesn’t yet tell us, is what this post is for.
HER2-Positive Breast Cancer and the Neoadjuvant Treatment Window
HER2 (human epidermal growth factor receptor 2) is a protein that promotes cell growth. In approximately 15 to 20% of breast cancers, the HER2 gene is amplified, producing too many HER2 receptors on tumor cell surfaces and driving aggressive cancer growth. HER2-positive breast cancer tends to grow faster than hormone receptor-positive cancer but is also more sensitive to HER2-targeted therapies.
Neoadjuvant therapy refers to systemic treatment given before surgery. This is different from adjuvant therapy, which is given after surgery to reduce recurrence risk. In HER2-positive early breast cancer, neoadjuvant treatment serves several purposes:
- It may downstage the tumor, reducing its size and lymph node involvement, potentially enabling less extensive surgery
- It allows oncologists to observe the tumor’s response to treatment in real time
- It provides important prognostic information that guides post-surgery treatment decisions
- Most critically: it creates the opportunity for pathologic complete response, the most meaningful outcome measure in this setting
| What is a pathologic complete response (pCR) and why does it matter? A pathologic complete response (pCR) means that when the removed tumor and lymph nodes are examined under a microscope after surgery, no viable invasive cancer cells are found. In DESTINY-Breast11, the pCR definition used was ypT0/is ypN0, meaning no residual invasive cancer in the breast (with allowance for non-invasive in-situ disease) and no cancer in the lymph nodes. pCR is one of the most important prognostic markers in HER2-positive breast cancer. Patients who achieve pCR have substantially lower rates of cancer recurrence and significantly better long-term survival than those with residual disease. The relationship between pCR and survival is why the FDA accepts it as a surrogate endpoint for approval in the neoadjuvant setting. The clinical decision implications extend beyond prognosis. Patients who achieve pCR typically continue with standard adjuvant therapy and may be candidates for less extensive surgery. Patients who do NOT achieve pCR are typically offered additional targeted therapy after surgery (currently, T-DM1/Kadcyla is the standard for residual HER2+ disease) to try to reduce their recurrence risk. Understanding pCR rates is therefore both a survival question and a treatment-planning question. |
|---|
What Enhertu Is and How It Works
Enhertu (trastuzumab deruxtecan, T-DXd) is an antibody-drug conjugate (ADC), a category of targeted therapy that links a cancer-targeting antibody to a chemotherapy payload. The antibody component is trastuzumab, which has been a cornerstone of HER2-positive breast cancer treatment for decades. The payload is deruxtecan, a topoisomerase I inhibitor chemotherapy. The linker between them is designed to be stable in the bloodstream but cleaved inside tumor cells.
The mechanism creates a guided delivery system. Trastuzumab finds tumor cells expressing HER2 on their surface and binds to them. The ADC is then internalized into the cell, where the linker is cleaved and the deruxtecan payload is released directly inside the cancer cell, causing it to die. A key additional property of T-DXd’s payload is what’s called a bystander effect: some of the released chemotherapy can diffuse into neighboring cancer cells, including those that may not strongly express HER2. This may help explain T-DXd’s activity even in heterogeneous tumors.
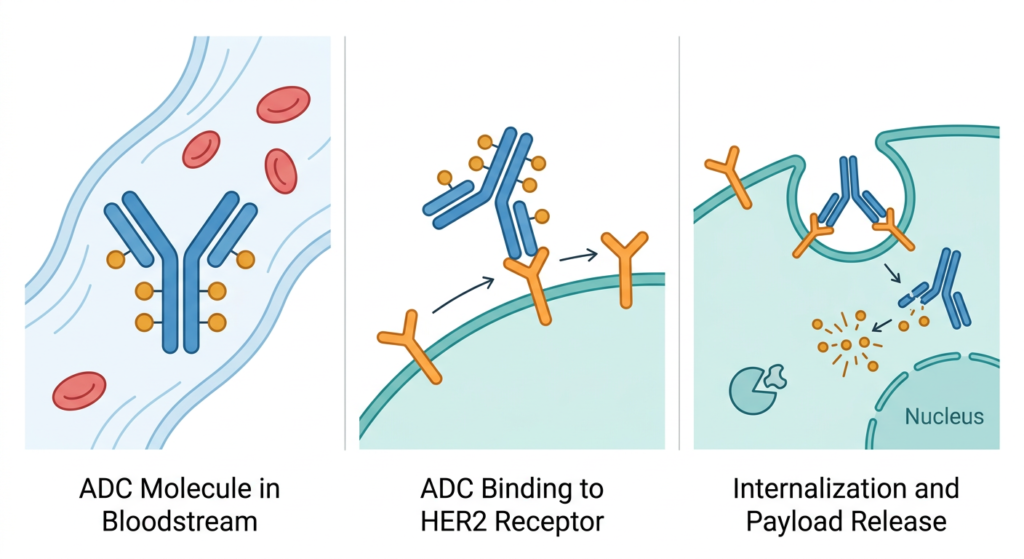
T-DXd already has FDA approvals for:
- HER2-positive metastatic breast cancer (second-line and beyond)
- HER2-low metastatic breast cancer (a broader category defined by lower HER2 expression)
- HER2-ultralow metastatic breast cancer
- HER2-positive metastatic gastric/gastroesophageal junction cancer
- HER2-mutant metastatic non-small cell lung cancer
If the May 18 decision is favorable, the neoadjuvant HER2-positive early breast cancer indication would be added to this list. For context on how ADC technology works in a related breast cancer setting, see our detailed coverage of Dato-DXd and TROPION-Breast02 in triple-negative breast cancer.
The DESTINY-Breast11 Trial: Design and Results
Design
DESTINY-Breast11 (NCT05113251) was a global, multicenter, randomized, open-label Phase 3 trial conducted at 147 sites across 18 countries. It enrolled adults with previously untreated, high-risk HER2-positive early breast cancer, defined as tumors that were either T3 or larger with any nodal status, or any T stage with N1 to N3 nodal involvement (node-positive disease), including inflammatory breast cancer. The HER2-positive definition required either IHC 3+ or positive in situ hybridization. Patients were randomized in a 1:1:1 design across three arms:
- T-DXd monotherapy arm: T-DXd 5.4 mg/kg every 3 weeks for 8 cycles (n=286), closed early
- T-DXd-THP arm: T-DXd 5.4 mg/kg every 3 weeks for 4 cycles, then paclitaxel plus trastuzumab plus pertuzumab (THP) for 4 cycles (n=321)
- ddAC-THP (control arm): Dose-dense doxorubicin plus cyclophosphamide every 2 weeks for 4 cycles, then THP for 4 cycles (n=320)
The T-DXd monotherapy arm was closed early in March 2024, following an Independent Data Monitoring Committee recommendation based on lower pCR rates than the combination arms and low likelihood of demonstrating superiority. This was a pre-specified adaptive design decision, not a safety signal.
Primary endpoint: pCR results
| Outcome | T-DXd plus THP (n=321) | ddAC plus THP (n=320) |
|---|---|---|
| pCR rate (ypT0/is ypN0) | 67.3% | 56.3% |
| Absolute difference in pCR | +11.0 percentage points | — |
| p-value | 0.003 | — |
| Patients proceeding to surgery | 97.2% | Comparable |
| EFS HR (immature, 4.5% events) | 0.56 (95% CI 0.26 to 1.17) | Reference |
Source: Harbeck N et al. Annals of Oncology. 2025. doi:10.1016/S0923-7534(25)04968-3. Presented at ESMO Congress 2025, Berlin. Abstract 291O.
The 11-point improvement in pCR rate is clinically meaningful by any standard in this disease. To put it in context: with existing standard-of-care regimens, pCR rates in high-risk HER2-positive disease range from approximately 39 to 64% depending on population characteristics and regimen. The DESTINY-Breast11 control arm (56.3%) sits in the middle of that range, reflecting an appropriately representative benchmark. The T-DXd-THP arm’s 67.3% is at the top of and above that historical range.
The benefit was consistent across pre-specified subgroups, including both hormone receptor-positive and hormone receptor-negative tumors, an important finding because HER2+/HR+ tumors historically have lower pCR rates and represent a more challenging treatment population.
What about long-term survival?
The EFS (event-free survival) hazard ratio of 0.56 suggests a 44% reduction in the rate of recurrence or death events in favor of T-DXd-THP numerically. However, EFS data maturity was only 4.5% at the time of the analysis, meaning very few events had occurred. The confidence interval (0.26 to 1.17) crosses 1.0, meaning the survival benefit is directionally promising but not yet statistically confirmed.
This is expected and appropriate for a neoadjuvant trial in early-stage cancer: these patients were diagnosed at a potentially curable stage, and survival events take years to accumulate. The FDA’s precedent for accepting pCR as a surrogate endpoint in the neoadjuvant setting means approval does not require mature survival data. More mature EFS and overall survival data from DESTINY-Breast11 will emerge over subsequent years and will be critical for confirming the long-term value of the pCR benefit.
Safety: Better Tolerability Than the Current Standard, With One Key Signal to Monitor
One of the most striking findings in DESTINY-Breast11 is not the efficacy. It is the safety comparison. The T-DXd-THP arm had substantially fewer severe adverse events than the anthracycline-based control.
| Safety Metric | T-DXd plus THP | ddAC plus THP |
|---|---|---|
| Grade 3 or higher adverse events | 37.5% | 55.8% |
| Serious adverse events | 10.6% | 20.2% |
| Treatment interruptions | 37.8% | 54.5% |
| Left ventricular dysfunction (all grade) | 1.3% | 6.1% |
| ILD/pneumonitis (all grade) | 4.4% | 5.1% |
| Grade 3/4 ILD events | 1 | 5 |
| Grade 5 (fatal) ILD events | 1 | 1 |
| Treatment-related deaths (all causes) | 1 (0.3%) | 2 (0.6%) |
| Most common Grade 3 or higher AE | Neutropenia (13.8%) | Hematologic toxicity predominant |
| Fatigue (all grade) | 41.3% | 54.8% |
The cardiac finding warrants specific emphasis. Doxorubicin (the “A” in AC chemotherapy) is associated with dose-dependent cardiotoxicity, including cardiomyopathy and heart failure, that can emerge during treatment and persist or worsen years later. The 6.1% rate of left ventricular dysfunction in the control arm versus 1.3% in the T-DXd-THP arm represents a clinically important difference in a population that will be living with the long-term consequences of treatment for decades.
| The ILD signal: the key safety consideration for T-DXd Interstitial lung disease (ILD), inflammation and scarring of lung tissue, is the most important safety concern with T-DXd across all its indications. In DESTINY-Breast11, the all-grade ILD rate was 4.4% in the T-DXd-THP arm and 5.1% in the control arm, which are comparable. Most events were Grade 1 or 2 and manageable with dose modification and corticosteroids. However, there was one Grade 5 (fatal) ILD event in each arm, with the independent adjudication committee attributing one death in the T-DXd-THP arm to drug-related pneumonitis. This is not a reason to avoid the drug; one in each arm is a roughly comparable rate at this sample size. But ILD monitoring is a critical clinical requirement for T-DXd in practice. Current guidance requires baseline pulmonary assessment before starting T-DXd, prompt evaluation of any new or worsening respiratory symptoms (dyspnea, cough, fever), immediate T-DXd interruption if ILD is suspected, and corticosteroid treatment for confirmed cases. Providers switching to this regimen must be familiar with ILD surveillance protocols. |
|---|
What This Means for Patients Navigating Treatment Right Now
If you have been diagnosed with HER2-positive breast cancer at stage II or III and are at the stage of discussing neoadjuvant treatment options with your oncologist, this FDA decision is directly relevant to your care.
If the FDA approves on May 18
- T-DXd followed by THP would become an FDA-approved option for high-risk (stage II/III) HER2-positive early breast cancer, specifically for patients with node-positive disease (N1 to N3) or large tumors (T3 or larger).
- Your oncologist may recommend this regimen over the current anthracycline-based standard, particularly if your tumor characteristics suggest high risk and you have cardiovascular risk factors that make doxorubicin’s cardiac effects a concern.
- The treatment involves 4 cycles of T-DXd (intravenous, every 3 weeks), followed by 4 cycles of paclitaxel plus trastuzumab plus pertuzumab, then surgery. Post-surgery treatment depends on whether you achieved pCR.
- If you have already started a neoadjuvant regimen: do not switch without discussion with your oncologist. Mid-treatment changes are complex and require careful individual assessment.
The treatment pathway after surgery
pCR does not mean treatment is finished. Patients who achieve pCR after neoadjuvant therapy typically continue with adjuvant trastuzumab with or without pertuzumab, and those with HR+ disease also receive endocrine therapy. Patients who do NOT achieve pCR currently receive adjuvant ado-trastuzumab emtansine (T-DM1/Kadcyla) to address the residual disease that proved resistant to neoadjuvant treatment.
A higher pCR rate means more patients entering that post-surgery phase in the most favorable prognostic position. For patients who still have residual disease, the adjuvant escalation pathway remains unchanged by this approval.
For related context on ADC mechanisms and how different ADCs work across breast cancer subtypes, see our post on vepdegestrant and the PROTAC mechanism in ESR1-mutated ER+ breast cancer and our analysis of Dato-DXd in triple-negative breast cancer.
Reading This Honestly: Context and Limitations
Survival data is immature
At 4.5% EFS maturity, we do not yet have confirmatory evidence that the pCR improvement translates into longer survival. The FDA’s precedent allows pCR as a surrogate, and the EFS directional signal (HR 0.56) is encouraging, but it is not yet proven. Patients and oncologists making decisions now are extrapolating from a strong surrogate, not from confirmed survival benefit. The maturing EFS and OS data from DESTINY-Breast11 will be the most important data to watch over the next several years.
Representation limitations
The published trial report specifically notes under-representation of Black or African American patients. This is a meaningful limitation in a disease where Black women are more likely to be diagnosed with aggressive subtypes and at advanced stages, and where outcomes disparities are well documented. Whether the pCR and safety results generalize fully to this population requires additional study and real-world evidence.
The monotherapy arm closed early
T-DXd monotherapy (without the THP sequence) achieved pCR rates of 43 to 51%, which is numerically inferior to both the combination arm and the control arm. The IDMC closed enrollment in that arm based on this finding. The approved regimen, if cleared, will be T-DXd followed by THP, not T-DXd alone. This distinction matters for clinical implementation.
What Happens on and After May 18
The PDUFA date is May 18, 2026. This is the deadline by which the FDA must complete its review. Decisions can come on or before this date.
If approved, T-DXd followed by THP would immediately be available to prescribers as the first ADC-based neoadjuvant regimen for HER2-positive early breast cancer. NCCN guideline updates and payer coverage decisions typically follow relatively quickly for priority FDA approvals, though individual insurance authorization timelines vary.
China has already approved this regimen based on the same DESTINY-Breast11 data. The European Medicines Agency review is ongoing. Regulatory validation across multiple agencies, if it follows, will strengthen the evidence base further.
We will update this post when the FDA’s ruling is announced.
Are you or a family member navigating a HER2-positive breast cancer diagnosis?
Treatment decisions in early breast cancer are among the most consequential and time-sensitive in oncology. If you have been recently diagnosed with HER2-positive stage II or III breast cancer, the most important step is consultation with a breast oncologist at a cancer center with expertise in HER2-directed therapies and access to current clinical trial data. The NCI-Designated Cancer Centers directory maintains a searchable list of specialized breast oncology programs. Susan G. Komen and the Metastatic Breast Cancer Alliance maintain patient navigation resources. We will continue tracking the FDA’s decision and the maturing DESTINY-Breast11 survival data as both become available.
Sources
PubMed: Harbeck N et al. DESTINY-Breast11. PubMed. PMID: 41130363.
ESMO 2025 abstract: Harbeck N et al. DESTINY-Breast11: Neoadjuvant T-DXd alone or followed by THP vs SOC for high-risk HER2+ eBC. ESMO Congress 2025. Abstract 291O.
DESTINY-Breast11 trial registration: NCT05113251. ClinicalTrials.gov.
AstraZeneca/Daiichi Sankyo press release: Enhertu followed by THP before surgery resulted in a pathologic complete response in 67% of patients in DESTINY-Breast11 Phase III trial. astrazeneca.com. October 18, 2025.
Targeted Oncology trial coverage: DESTINY-Breast11: Neoadjuvant T-DXd/THP Improves pCR in High-Risk HER2+ BC. targetedonc.com. October 2025.
FDA surrogate endpoint resource: Surrogate Endpoint Resources for Drug and Biologic Development. FDA.gov.
T-DM1 FDA approval: FDA approves ado-trastuzumab emtansine for HER2-positive breast cancer. FDA.gov.
Patient resources: NCI Cancer Center directory | Susan G. Komen | MBC Alliance | NCCN Breast Cancer Guidelines
| Disclaimer: Health Evidence Digest provides general information about clinical trials and FDA regulatory processes for educational purposes. This content is not a substitute for professional medical advice, diagnosis, or treatment. Decisions about breast cancer treatment, including neoadjuvant therapy, should be made in close consultation with a qualified oncologist who can account for your individual diagnosis, tumor characteristics, and health status. |
|---|