
| 📌 The essentials On April 23, 2026, the FDA approved Otarmeni (lunsotogene parvec-cwha, Regeneron) as the first FDA-approved gene therapy for inherited deafness in history. The therapy is indicated for children and adults with profound hearing loss due to biallelic mutations in the OTOF gene, which causes a condition where the inner ear is structurally normal but cannot transmit sound signals to the brain. The clinical basis: Results from the CHORD Phase 1/2 trial (NCT05295056) showing 80% of participants (16 of 20) achieved or exceeded the primary endpoint at 6 months, and 42% of participants with longer follow-up achieved normal hearing. Nine of 12 children who received the therapy gained enough hearing to stop using cochlear implants. The approval was granted under accelerated approval with continued approval contingent on confirmatory trial results. This was also the first gene therapy approved under the FDA’s Commissioner’s National Priority Voucher (CNPV) program, approved in just 61 days after BLA submission. The price: Regeneron has stated it will provide Otarmeni at no cost for the drug itself to eligible patients in the United States. Important caveat: the surgical procedure required to administer it is not covered by Regeneron and will be subject to normal insurance and cost-sharing. |
|---|
When Travis Smith was born, he failed his newborn hearing test. His mother, Sierra, was told it was probably just fluid in the ears. But weeks passed, and nothing changed. Slamming pots and pans, yelling his name — nothing reached him. Travis was, as Sierra later described it, 100% deaf.
A few months later, after genetic testing confirmed a mutation in a gene called OTOF, Travis received an experimental treatment at Columbia University in New York. About ten weeks after the procedure, Sierra laughed loudly while driving. Travis, asleep in his car seat, startled for the first time. She and her friend started yelling. He woke up.
On April 23, 2026, that experimental treatment became Otarmeni (lunsotogene parvec-cwha), the first FDA-approved gene therapy for inherited deafness in history. And in a move that surprised nearly everyone in the pharmaceutical industry, Regeneron announced it will provide the drug at no cost to eligible patients in the United States.
There is a lot to unpack here: the science, the price, the very reasonable counterarguments from the Deaf community, and what this means for the larger field of genetic hearing loss.
What Is OTOF-Related Hearing Loss?
Hearing happens through a remarkably precise chain of events. Sound waves enter the ear canal, cause the eardrum to vibrate, and those vibrations travel through three tiny bones in the middle ear before reaching the cochlea, the snail-shaped structure of the inner ear. Inside the cochlea, thousands of hair cells convert those vibrations into electrical signals. A protein called otoferlin is what allows those hair cells to release the neurotransmitters that carry those signals to the auditory nerve and then on to the brain.
In children with biallelic mutations in the OTOF gene, meaning they inherited a non-working copy from both parents, otoferlin is absent or non-functional. The cochlea is structurally intact. The hair cells are there. Sound waves are converted normally. But the signal cannot be passed to the brain because the neurotransmitter release mechanism is broken. The result is profound sensorineural deafness from birth, despite an otherwise normal-looking inner ear.
OTOF mutations account for roughly 2% to 8% of inherited non-syndromic hearing loss, according to the FDA. In absolute numbers, about 50 babies are born each year in the United States with the condition, a number small enough that most audiologists and pediatricians will rarely encounter it. But the impact on those families is total..
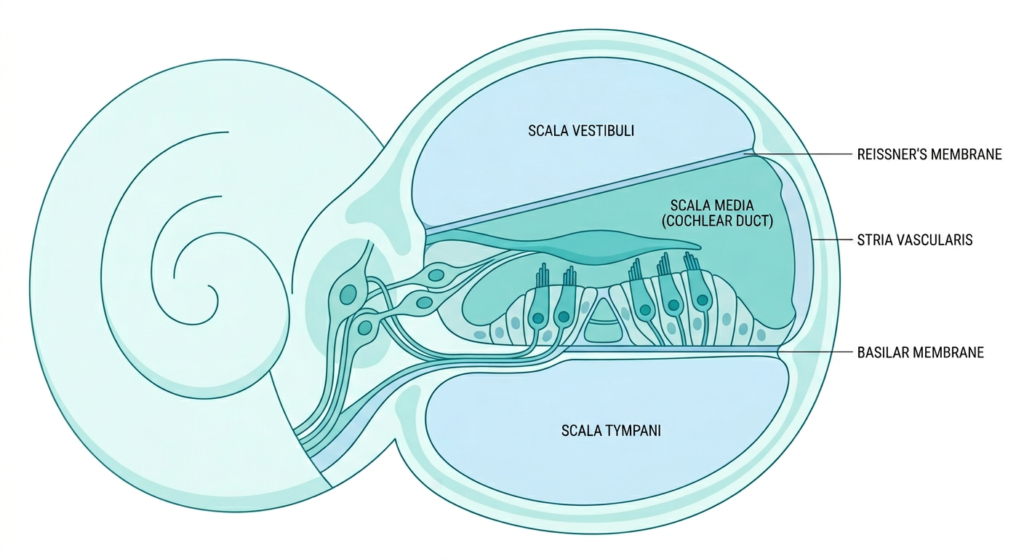
How Otarmeni Works
Otarmeni is an adeno-associated virus (AAV) vector-based gene therapy, specifically a dual-vector system, because the OTOF gene is unusually large and too big to fit inside a single AAV. Regeneron’s approach splits the gene in half across two AAV serotype 1 vectors that are co-administered. Once inside the hair cells, the two halves recombine to produce a functional OTOF gene, which then directs the cells to make working otoferlin protein.
The treatment is administered surgically. Under general anesthesia, a surgeon makes a small incision behind the ear to access the cochlea and delivers the viral vectors directly into the fluid-filled space of the inner ear via a syringe and catheter, a procedure similar in approach to cochlear implant surgery, though the anatomy targeted is slightly different. The therapy can be given to one ear or both.
One important technical detail: the OTOF gene in Otarmeni is under the control of a proprietary Myo15 promoter, which is designed to restrict gene expression specifically to hair cells that normally produce otoferlin. This cell-type specificity is important both for efficacy and safety, as it reduces the chance of off-target expression in tissues that do not need the protein.
| Why is the OTOF gene so large, and why does that matter? Standard single-AAV gene therapies are limited by the packaging capacity of the virus, roughly 4.7 kilobases of genetic material. The OTOF gene is approximately 6 kilobases, which has long made it technically challenging to deliver in a single vector. Regeneron’s dual-AAV approach is one of several strategies the field has developed to work around this constraint. It addresses the same large-gene delivery challenge that has been encountered in gene therapy for conditions like Duchenne muscular dystrophy. The fact that this approach produced consistent, durable results in the CHORD trial is a meaningful technical achievement, not just for hearing loss, but for the broader field of large-gene delivery. |
|---|
The CHORD Trial: What the Clinical Data Shows
The FDA approval is based on results from the CHORD trial (NCT05295056), an ongoing, registrational Phase 1/2 multicenter, open-label study. Twenty participants aged 10 months to 16 years with molecularly confirmed OTOF mutations received a single dose of Otarmeni in one or both ears. The primary endpoint was improvement in hearing sensitivity measured by pure-tone audiometry at week 24.
| CHORD trial key results | |
|---|---|
| Participants meeting or exceeding primary endpoint at 6 months | 16 of 20 (80%) |
| Participants achieving normal hearing with longer follow-up | 42% |
| Children who stopped using cochlear implants after treatment | 9 of 12 |
| Minimum follow-up with durable hearing benefits | At least 2 years |
| Age range in trial | 10 months to 16 years |
| Effect of age at treatment on efficacy | Not significant, which supported label inclusion of adults |
| Most common adverse events | Middle ear infection or inflammation, vomiting, nausea, dizziness (consistent with surgical procedure) |
Source: CHORD Phase 1/2 trial, NCT05295056. Primary results published in NEJM, 2026.
| Accelerated approval: what it means here Otarmeni received accelerated approval based on improvement in pure-tone audiometry as a surrogate endpoint. Continued approval may be contingent upon verification of treatment effects on clinical measures of speech development and quality of life, the outcomes families ultimately care most about. The confirmatory portion of the CHORD trial is ongoing. The FDA specifically notes that durability of hearing improvement is a key variable still being assessed. For a one-time gene therapy, how long the benefit lasts is the central question that will define long-term clinical value and public health cost-effectiveness. The approval was also notably fast: granted just 61 days after the Biologics License Application was filed, tied for the fastest BLA approval in modern FDA history, and the first gene therapy approved under the FDA’s Commissioner’s National Priority Voucher (CNPV) program. For context on how the CNPV program works and which other drug programs have received vouchers, see our post on the FDA’s fast-tracking of three psychedelic drug programs. |
|---|
The Price Tag: $0. What Is Actually Going On There?
Gene therapies for rare diseases are expensive. Not slightly expensive — the kind of expensive that regularly makes headlines. Hemgenix (hemophilia B) was priced at $3.5 million per patient. Zolgensma (spinal muscular atrophy) at $2.1 million. Casgevy (sickle cell disease) at $2.2 million. These prices reflect the reality of developing treatments for patient populations sometimes numbering in the hundreds, where there is no scale to amortize development costs.
Regeneron’s internal analysis suggested Otarmeni could have been priced as high as $4 million per patient, generating an estimated $200 million to $400 million in annual revenue. The company chose not to. Regeneron’s co-founder and president, Dr. George Yancopoulos, acknowledged the company made a deliberate choice to prioritize access over revenue from this particular therapy, despite internal discussion about alternative pricing models.
That decision came alongside Regeneron’s participation in the Trump administration’s Most Favored Nation drug pricing announcement, a policy effort to bring U.S. drug prices more in line with prices paid in European and Asian markets. The timing was politically convenient, but the substance of offering the therapy free stands regardless of the surrounding context.
Sarah Emond, President and CEO of the Institute for Clinical and Economic Review (ICER), noted in a statement following the approval that Regeneron has shown that one option companies can consider to ensure affordable patient access to these therapies is to simply not charge the health system for the drug. She called it a model worth understanding for what it demonstrates about the range of approaches available to developers of rare disease therapies.
There are important nuances in the “free” framing worth noting clearly. Regeneron is providing the drug itself at no cost to clinically eligible patients. The company does not control and is not covering the cost of the surgical procedure required to administer it. Cochlear implant surgery, which uses a similar approach, typically costs between $30,000 and $100,000 including hospitalization and anesthesia. The out-of-pocket portion for patients will depend on their insurance coverage for the procedure, not the drug.
Otarmeni’s pricing model also has no established precedent for international markets. CEO Leonard Schleifer told CNBC that overseas pricing has not been set, stating that other countries should pay their fair share. For families outside the United States with children who have OTOF mutations, the picture is much less clear.
A Perspective Worth Sitting With: The Deaf Community Response
Not everyone greeted this approval with unqualified celebration, and that response deserves more than a footnote.
Jaipreet Virdi, a historian of medicine, technology, and deafness at the University of Victoria who is herself deaf, raised a concern that has been articulated within Deaf culture for years: that genetic therapies targeting deafness can reinforce the assumption that deafness is a deficiency to be corrected rather than a difference to be accommodated. For members of the Deaf community who use sign language, have Deaf cultural identities, and live full, rich lives, a medical framing of deafness as a problem in need of eradication is not a neutral position.
This is not a fringe view. It is a well-established strand of Deaf cultural identity that preceded cochlear implants and will continue to evolve as genetic therapies expand. It does not invalidate what Otarmeni has done for Travis, or Miles, or the other children in the CHORD trial. But it does mean that the conversation around who benefits from these therapies, and on what terms, is more complex than the headline numbers suggest.
Regeneron’s own press release acknowledged this directly. Janet DesGeorges, Executive Director of Hands and Voices, a family-driven organization supporting children with all forms of hearing loss and all communication approaches, was quoted in the approval announcement noting that families deserve access to balanced information and a range of options when navigating genetic hearing loss, and that the choice of approach belongs to individual families.
| Cochlear implants versus gene therapy: how they are different Cochlear implants are electronic devices surgically implanted in the inner ear that bypass damaged hair cells and directly stimulate the auditory nerve. They restore useful hearing for many patients but do not restore physiological hearing. The sound quality is different from natural hearing and varies considerably between users. They require external processors worn behind the ear, run on batteries, and must be managed over a lifetime. Otarmeni, by contrast, restores the biological mechanism of hearing by enabling the hair cells themselves to function. The hearing it produces is closer to natural hearing that is present continuously without external hardware. However, it only works for patients with OTOF mutations who have no prior cochlear implant in the ear to be treated. The two approaches are not directly comparable and serve partially overlapping but distinct populations. |
|---|
Beyond OTOF: What This Approval Unlocks
OTOF mutations account for only 1% to 3% of cases of genetic hearing loss at birth. The significance of this approval is therefore less about its immediate patient population, roughly 50 children per year in the U.S., and more about what it proves and where it leads.
Genetic hearing loss involves more than 100 identified genes. OTOF attracted early attention because its mechanism was well-understood, the hair cell pathology is isolated (outer hair cell function is preserved), and the AAV delivery route to the cochlea had been mapped in preclinical models. Proving that this approach works, that you can deliver a gene to inner ear hair cells via surgical infusion and produce durable, functional hearing, is the foundational result the broader field needed.
Eli Lilly and several academic groups are also developing gene therapies targeting OTOF, many showing comparably strong results. The publication of strong data in the New England Journal of Medicine in 2026, which preceded and contributed to the FDA’s accelerated review, has drawn significant investment into the broader genetic hearing loss space. Dr. Lawrence Lustig of Columbia University, who treated several CHORD participants, noted substantial interest in pursuing other forms of genetic deafness that are more common, and that investment is now arriving.
Researchers are also beginning to consider whether someday gene therapy approaches might address acquired hearing loss from aging or noise exposure, which affects hundreds of millions of people globally. That is a much longer road, requiring different targets and delivery methods. But the clinical validation of cochlear gene delivery in OTOF patients makes it a more credibly walkable path than it was before April 23, 2026.
What This Approval Does Not Yet Answer
How long does the benefit last?
The CHORD trial has follow-up of at least two years in some participants, and hearing benefits have been durable over that period. But two years is a short window for what is being offered as a one-time, potentially permanent treatment, particularly for children who may live for seven more decades. Long-term follow-up from the confirmatory CHORD trial will be critical. The FDA has specifically listed durability of hearing improvement as a condition of continued approval.
What about speech and language development?
Pure-tone audiometry tells us whether a patient can detect sounds at various frequencies and volumes. It does not directly measure what matters most to families: speech comprehension, language acquisition, and the ability to communicate in the ways they choose. The confirmatory trial is tasked with verifying treatment effects on these clinical measures. The gap between “can detect a whisper” and “is developing speech and language normally” is the one families and clinicians most need filled.
Which patients are candidates?
The indication requires molecularly confirmed biallelic OTOF variants, preserved outer hair cell function (confirmed by otoacoustic emissions testing), and no prior cochlear implant in the ear to be treated. Genetic testing infrastructure for identifying OTOF mutations in newborns varies considerably across health systems. The therapy’s real-world reach will depend partly on how systematically genetic diagnosis of congenital deafness is pursued, which is currently inconsistent in the U.S.
For families navigating genetic hearing loss:
This approval touches on intersecting questions: the science of gene delivery, the ethics of treating deafness, the unprecedented pricing decision, and what proof-of-concept in OTOF means for the dozens of other genetic causes of hearing loss. For families with children recently diagnosed with genetic hearing loss, regardless of which gene is involved, several organizations maintain current resources:
Hands and Voices supports families navigating all communication approaches without advocacy for any single one. The National Association of the Deaf (NAD) provides resources from a Deaf cultural perspective. The Hearing Loss Association of America (HLAA) offers advocacy and practical support resources. The NIDCD maintains clinical information on cochlear implants and emerging therapies. Families interested in the CHORD confirmatory trial or other OTOF gene therapy studies can search for open enrollment studies at ClinicalTrials.gov.
Sources
FDA approval announcement: FDA Approves First-Ever Gene Therapy for Treatment of Genetic Hearing Loss Under National Priority Voucher Program. FDA.gov. April 23, 2026.
Regeneron press release: Otarmeni (lunsotogene parvec-cwha) Approved by FDA. investor.regeneron.com. April 23, 2026.
CHORD trial registration: NCT05295056. ClinicalTrials.gov.
Primary clinical data: CHORD Phase 1/2 trial results. New England Journal of Medicine. 2026.
ICER pricing commentary: Institute for Clinical and Economic Review. Statement on Otarmeni pricing. icer.org.
Pricing context (CNBC): Schleifer L. Regeneron weighs overseas price for Otarmeni. CNBC. April 24, 2026.
Deaf community perspective: Virdi J. Quoted in NPR/KERA News. Rob Stein. The FDA gives the green light to the first gene therapy for deafness. keranews.org. April 23, 2026.
Hands and Voices: handsandvoices.org. Cited in Regeneron approval press release.
Patient story (Travis): NPR/KERA News. Rob Stein. April 23, 2026.
Patient story (Miles): CNN. Meg Tirrell. April 23, 2026.
Pipeline context: Gene therapy for deafness approved. Science. April 23, 2026.
Patient and family resources: Hands and Voices | National Association of the Deaf | Hearing Loss Association of America | NIDCD Cochlear Implants | ClinicalTrials.gov: OTOF hearing loss
| Disclaimer: Health Evidence Digest provides general information about health research and FDA decisions for educational purposes. This content is not a substitute for professional medical advice, diagnosis, or treatment. Accelerated approval does not constitute final confirmation of clinical benefit. The confirmatory CHORD trial is ongoing. Always consult a qualified audiologist, otolaryngologist, or geneticist regarding treatment decisions for your child or yourself. |
|---|
Leave a Reply