| 📌 What changed and when January 16, 2026: The FDA approved a supplemental NDA extending Nexplanon’s approved duration from 3 years to 5 years. February 23, 2026: New REMS (Risk Evaluation and Mitigation Strategy) program launched. Nexplanon is now only available through REMS-enrolled providers. August 23, 2026: Deadline by which all providers who insert or remove Nexplanon must complete REMS certification or lose ordering access. If you have a Nexplanon currently: it may now be left in place for up to 5 years from insertion, rather than 3. Discuss your specific timeline with your provider. |
|---|
If you have a Nexplanon implant in your arm, there’s a good chance you’ve heard the three-year date coming up like a countdown. Get it out, replace it, or switch to something else. That schedule just changed.
On January 16, 2026, the FDA approved a supplemental new drug application from Organon extending Nexplanon’s approved duration of use from three years to five years. The approval is based on a clinical trial that followed 399 women through years four and five of continuous implant use and found zero pregnancies and no new safety findings. The same implant, the same arm, two additional years of coverage.
This post covers the clinical evidence behind the extension, what the new REMS program means for patients and providers, who can benefit from the extended duration and who should still remove at three years, and what to do if you have an implant right now.
What Nexplanon Is and How It Works
Nexplanon is a single-rod subdermal contraceptive implant, a small, flexible rod about 4 centimeters long and 2 millimeters wide, roughly the size of a matchstick. It is inserted just under the skin of the inner, non-dominant upper arm by a trained provider. It contains 68 mg of etonogestrel, a synthetic progestin, along with 15 mg of barium sulfate (which makes the implant visible on X-ray) and 0.1 mg of magnesium stearate.
Etonogestrel is released slowly and continuously from the implant into the bloodstream. It prevents pregnancy through three complementary mechanisms:
- Suppression of ovulation: the primary mechanism. Etonogestrel inhibits the LH surge that triggers ovulation. No egg released means no pregnancy possible.
- Thickening of cervical mucus: even if ovulation were to occur, thickened cervical mucus creates a barrier that sperm cannot penetrate effectively.
- Modification of the endometrium: changes in the uterine lining create an additional unfavorable environment for implantation.
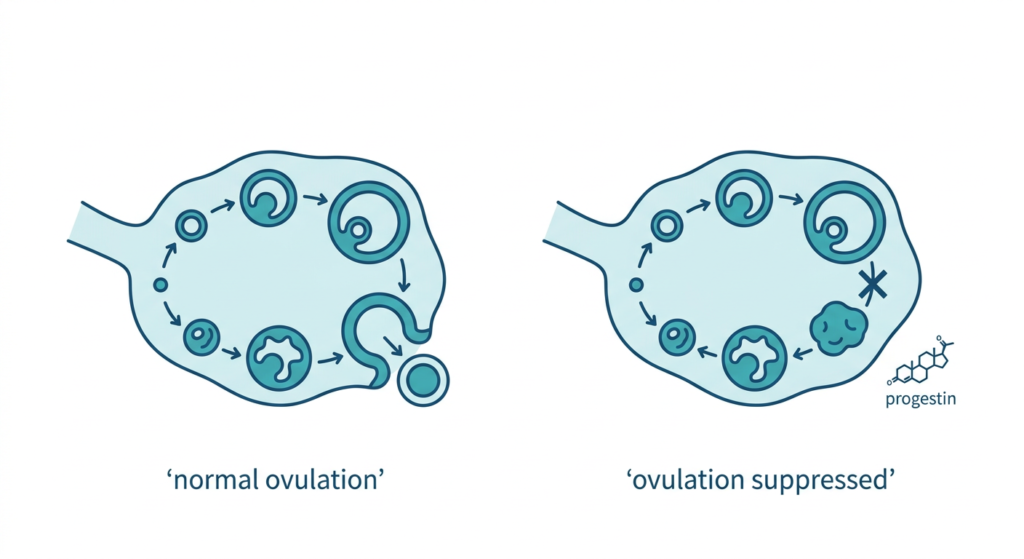
These three mechanisms together make Nexplanon one of the most effective contraceptive methods available. Its failure rate is less than 0.1% per year, lower than tubal ligation, vasectomy, IUDs, pills, patches, rings, and condoms. Unlike daily pills or monthly rings, it works without any ongoing action on the patient’s part: once inserted, it simply works.
It is also fully reversible. Fertility returns rapidly after removal, typically within days to weeks, not months. This makes it suitable for patients who want highly effective contraception now but may want to conceive in the future.
The Clinical Evidence Behind the 5-Year Extension
The FDA’s extension is based on data from a dedicated clinical trial designed specifically to evaluate contraceptive efficacy and safety during years four and five of use, the period for which no prior approval existed.
Study design
NCT04626596 was a multicenter, single-arm, open-label study conducted in the United States. It enrolled 399 women aged 18 to 35 who had been using a Nexplanon implant for exactly 36 months (within a two-week window). Participants continued using the same implant for an additional 24 months, with visits at months 6, 12, 18, and 24 of the extended period. The primary efficacy endpoint was the Pearl Index during years four and five.
The results
| Measure | Result |
|---|---|
| Participants enrolled | 399 women aged 18 to 35 (mean age 27) |
| Pregnancies during years 4 to 5 | Zero |
| Pearl Index (years 4 to 5) | 0.0 per 100 women-years (95% CI 0.00 to 0.69) |
| Mean BMI | 29.4 kg/m² (range 17.2 to 64.3) |
| Participants with BMI 30 or above (obesity) | 152 participants, 38.1% |
| Participants with BMI 40 or above (severe obesity) | 40 participants, 10.0% |
| New safety findings in years 4 to 5 | None |
| Safety profile vs. years 1 to 3 | Consistent; no new or worsening signals |
| Most common adverse event (extended use) | Intermenstrual (irregular) bleeding |
Source: NCT04626596. Organon press release January 16, 2026. FDA sNDA approval January 16, 2026.
The Pearl Index of 0.0, meaning zero pregnancies observed per 100 women-years of use, is the strongest possible contraceptive efficacy result. The upper bound of the 95% confidence interval (0.69) confirms that even accounting for statistical uncertainty, the maximum plausible pregnancy rate during years four and five remains well below 1 per 100 women-years. This is consistent with the established efficacy in years one through three.
| The BMI data is important and clinically underappreciated Weight-based concerns about hormonal contraceptive efficacy are a common patient question. Higher body weight means more body fat, and progestin hormones can be distributed into fat tissue, potentially lowering circulating hormone levels. For some hormonal methods, particularly emergency contraception and some combined pills, this is a documented efficacy concern. For the etonogestrel implant, the clinical trial data has consistently shown maintained efficacy across a wide BMI range. The year 4 to 5 extension study enrolled participants with a mean BMI of 29.4, nearly at the obesity threshold, with a full 38% above BMI 30 and 10% above BMI 40. Zero pregnancies across this population is a meaningful data point for clinicians advising patients with obesity on contraceptive options. The updated label explicitly reflects this diversity. |
|---|
Who Benefits From the Extended Duration and Who Should Still Remove at 3 Years
The 5-year approval is a maximum, not a mandate. Every patient’s situation is individual, and the right duration depends on goals, health status, and preferences.
Patients likely to benefit from extending to 5 years
- Those who are satisfied with their current implant experience and have no plans to conceive in the next two years
- Those who want to avoid an additional insertion/removal procedure, a minor but real burden for some patients
- Those with irregular access to healthcare where scheduling a replacement at exactly 3 years is difficult
- Those for whom cost is a factor: one fewer procedure and one fewer device every 5 years versus every 3
Patients who may prefer removal at or before 3 years
- Those planning pregnancy in the next year or two
- Those experiencing side effects (irregular bleeding, headache, mood changes, weight changes) that are troublesome and haven’t improved
- Those who want to switch to a different contraceptive method
- Those who have significant medical changes since insertion that affect contraceptive choice or introduce new contraindications
If You Have a Nexplanon Right Now: What This Means for You
This is the question most current implant users will want answered directly.
My implant is approaching or just past 3 years
The FDA approval means you now have the option to leave your existing implant in place for up to 5 years from the original insertion date, not 5 more years from now. Contact your provider to discuss whether extending is right for you. If you and your provider decide to extend, no procedure is needed; you simply continue as-is. If your implant has already been in place for more than 3 years and you were unaware of the extension, discuss with your provider promptly. The implant must be removed by the end of year 5.
My implant was recently inserted (within the last year or two)
When you approach the previously expected 3-year mark, you can now choose to stay with your current implant for an additional 2 years, as long as you remain a good candidate for continued use and have no contraindications at that point. No action needed now.
My implant was removed and replaced recently, or I’m planning removal
If you had your implant replaced at or before 3 years, you cannot retroactively extend the old implant. That device has been removed. Your new implant can now be used for up to 5 years from its insertion date. If you scheduled removal before learning about the extension and want to delay, contact your provider’s office to discuss.
The New REMS Program: What It Is and Why It Exists
Alongside the duration extension, the FDA required implementation of a new Risk Evaluation and Mitigation Strategy (REMS) for Nexplanon. A REMS is a safety program the FDA uses when a product has known or potential risks that require specific measures beyond standard labeling to manage safely. Nexplanon’s REMS exists specifically to address the risk of complications from improper insertion and removal.
Why improper insertion is a real concern
Nexplanon insertion is a minor procedure, but it is a procedure, and getting it wrong has consequences. An implant placed too deeply (intramuscular or beyond) can be difficult or impossible to remove without surgery. Migration, where the implant or a fragment moves from the insertion site, has been reported and sometimes requires more complex removal. Insertion near neurovascular structures can cause nerve damage. The REMS is designed to ensure that every provider who offers Nexplanon has the training to place it correctly, at the recommended depth and location, every time.
What the REMS requires for providers
| Step | What’s Required |
|---|---|
| 1. Register at nexplanonrems.com | Create an account at the REMS portal |
| 2. Review the Healthcare Provider Guide | Read through the updated training materials |
| 3. Review the Prescribing Information | Updated to reflect 5-year duration and BMI data |
| 4. Complete Knowledge Assessment | 7-question online quiz (~10 minutes total for steps 1 to 4) |
| 5. Complete Enrollment Form | Submit to receive REMS certification |
| 6. Complete in-person practical training | Required if not previously Organon-trained; includes competency checklist |
| 7. Report insertion/removal events | Any complications must be reported using the IRRE form or by calling 1-833-NXP-REMS |
| Critical provider deadline: August 23, 2026 Providers who insert or remove Nexplanon must complete REMS certification by August 23, 2026. After this date, providers who have not enrolled will not be able to order or receive Nexplanon for insertion. The enrollment window opened February 23, 2026. For providers who completed Organon’s previous Clinical Training Program, the online REMS enrollment (steps 1 to 5 above) may be sufficient without additional in-person training, depending on whether training history is reflected in the portal. If the system indicates training is required despite prior completion, call 1-833-NXP-REMS (1-833-697-7367). There are no changes to the recommended insertion technique or location. The correct site remains 3 to 5 cm posterior to the sulcus between the biceps and triceps on the inner upper arm. Full guidance is available at nexplanonrems.com. |
|---|
For patients: the REMS is a provider-side requirement. You do not need to do anything differently. It does mean that your provider must be REMS-certified to insert or remove your implant. If you are concerned about whether your current provider is enrolled, you can ask them directly or contact Organon’s REMS support line at 1-833-697-7367.
Safety, Side Effects, and Contraindications
Common side effects (from years 1 to 3 trials)
These were reported in at least 5% of clinical trial participants and remain the primary side effects to counsel patients about:
- Headache (24.9%), the most common
- Vaginitis (14.5%)
- Weight increase (13.7%)
- Acne (13.5%)
- Breast pain (12.8%)
- Abdominal pain (10.9%)
- Pharyngitis/upper respiratory symptoms (10.5%)
- Changes in menstrual bleeding patterns (the most common reason for discontinuation, at 11.1%)
Menstrual pattern changes deserve specific mention because they are common and often unexpected. Nexplanon can cause irregular spotting or light bleeding, more frequent bleeding, less frequent bleeding, or complete absence of periods, and the pattern that develops in the first three months is broadly predictive of what to expect going forward. This is not a sign of pregnancy or a problem; it is a normal pharmacological effect of progestin on the endometrium. For patients who find their bleeding pattern unacceptable, removal and method change remains an option at any time.
Contraindications: who should not use Nexplanon
Per the updated prescribing information, Nexplanon is contraindicated in patients with:
- Known or suspected pregnancy
- Current or past history of thrombosis or thromboembolic disorders
- Liver tumors (benign or malignant) or active liver disease
- Undiagnosed abnormal uterine bleeding
- Known or suspected breast cancer, personal history of breast cancer, or other progestin-sensitive cancer
- Allergy to any component of Nexplanon
Drug interactions to know
Certain medications that induce the CYP3A4 enzyme, including rifampin, some anti-seizure medications (carbamazepine, phenytoin, phenobarbital), and St. John’s Wort, can accelerate etonogestrel metabolism and potentially reduce implant efficacy. Patients starting these medications should discuss contraceptive options with their provider. The implant does not interact with most common medications.
Cost and Insurance Coverage
Under the Affordable Care Act, most private insurers are required to cover FDA-approved contraceptive methods without cost-sharing, meaning $0 out-of-pocket for the patient, including the device and the insertion procedure. This applies to Nexplanon. However, coverage details vary by plan, and some plans have grandfathered exemptions or denominational exemptions. It is always worth verifying with your specific insurer.
For uninsured or underinsured patients, the 5-year duration represents meaningful cost savings, one fewer device and insertion procedure per 10 years of use compared to the 3-year schedule. Title X family planning clinics provide Nexplanon on a sliding-scale fee basis. Organon’s patient support program can assist with coverage navigation; contact information is available at organon.com.
This extension is part of a broader pattern of evidence-based updates improving access to women’s reproductive healthcare in 2026. For related coverage, see our post on new 2026 cervical cancer self-collection screening guidelines and our overview of GLP-1 medications and PCOS fertility research.
Do you have questions about your implant or your timeline?
The FDA’s 5-year extension is a straightforward evidence-based update: the implant keeps working, the safety profile stays consistent, and women with BMI across a wide range continue to be protected. For women with a current implant approaching the 3-year mark, the conversation with your provider is now richer; you have a real choice rather than an automatic expiration date. For providers: the REMS enrollment deadline of August 23, 2026 is actionable now. The enrollment process at nexplanonrems.com takes approximately 10 minutes for those who have completed prior training. Bedsider’s provider resource page and the Reproductive Health Access Project have step-by-step REMS guides for clinical teams.
Sources
Organon press release: Organon Announces FDA Approval of sNDA Extending Duration of Use of NEXPLANON (etonogestrel implant) 68 mg Radiopaque. January 16, 2026.
FDA updated prescribing information: NEXPLANON Prescribing Information (2026). accessdata.fda.gov.
Clinical trial registration: NCT04626596: Study to Assess Contraceptive Efficacy and Safety of ENG Implant Beyond 3 Years of Use. ClinicalTrials.gov.
Contemporary OB/GYN: FDA approves 5-year use for etonogestrel implant 68 mg contraceptive. contemporaryobgyn.net. January/March 2026.
ReproHH (UCSF): FDA Approves Updated Nexplanon Label and Launches New REMS: What to Know. reprohh.ucsf.edu. February 2026.
Reproductive Health Access Project: Contraceptive Pearl: New FDA REMS Requirement on Nexplanon. reproductiveaccess.org. March 2026.
Bedsider for providers: Nexplanon REMS Requirements: What Providers Need to Know. providers.bedsider.org. March 2026.
Organon Pro (REMS FAQs): FAQs: NEXPLANON for HCPs. organonpro.com.
REMS enrollment: nexplanonrems.com. REMS support: 1-833-NXP-REMS (1-833-697-7367)
ACOG LARC resource: Long-Acting Reversible Contraception: Intrauterine Device and Implant. ACOG.
| Disclaimer: Health Evidence Digest provides general information about FDA approvals and health research for educational purposes. This content is not a substitute for professional medical advice. Decisions about contraceptive methods, including whether to extend Nexplanon use to 5 years, should be made in consultation with a qualified healthcare provider who can account for your individual health history and circumstances. |
|---|