
| 📌 The essentials On June 8, 2026, the FDA approved an expanded indication for Hympavzi (marstacimab-hncq, Pfizer) to include two additional patient populations: patients aged 12 years and older with hemophilia A or B who have inhibitors, and pediatric patients aged 6 to 11 years with hemophilia A or B with or without inhibitors. Hympavzi was originally approved in October 2024 for patients aged 12 years and older without inhibitors. The complete current indication: routine prophylaxis to prevent or reduce bleeding episodes in adults and pediatric patients aged 6 years and older with hemophilia A (congenital factor VIII deficiency) or hemophilia B (congenital factor IX deficiency), with or without inhibitors. First-in-class designations from the expanded approval: Hympavzi is now the first subcutaneous non-factor therapy available for children aged 6 to 11 years with hemophilia B. Mechanism: Hympavzi targets the Kunitz 2 domain of tissue factor pathway inhibitor (TFPI), a natural anticoagulant that brakes the initiation of coagulation. By inhibiting TFPI, marstacimab restores the coagulation pathway in a way that works regardless of whether the patient is deficient in factor VIII or factor IX, and regardless of whether inhibitory antibodies are present against those factors. This is why it works in both hemophilia A and B, and in patients with inhibitors. The clinical basis: Phase 3 BASIS trial (NCT03938792) inhibitor cohort and Phase 3 BASIS KIDS trial (NCT05611801). BASIS inhibitor cohort: 93% reduction in mean treated annualized bleeding rate (ABR) versus on-demand bypassing agent therapy (1.4 [95% CI 0.9 to 2.3] versus 19.8 [95% CI 16.1 to 24.3]; ratio 0.07 [95% CI 0.04 to 0.12]; p less than 0.0001). BASIS KIDS (ages 6 to 17): mean ABR 1.8 versus historical model-based mean ABR 3.6 with prior routine prophylaxis. Pediatric inhibitor subgroup (ages 6 to under 18; n=14): mean ABR 1.4 (99% CI 0.5 to 4.5) versus historical mean ABR 18.9 (99% CI 14.2 to 25.2). Ages 6 to 11 cohort (n=7): mean ABR 1.3 (99% CI 0.5 to 3.4). Warnings: thromboembolic events occurred in 2 of 259 patients in the open-label extension study; hypersensitivity reactions; embryo-fetal toxicity; increased laboratory values of fibrin D-dimer and prothrombin fragment 1.2. Dosing: once weekly subcutaneous injection; available as 75 mg/0.5 mL and 150 mg/mL prefilled syringe or autoinjector pen. No routine treatment-related laboratory monitoring required. |
|---|
Hemophilia is, at its core, a disease of imbalance. The coagulation cascade that allows blood to clot and stop bleeding is a tightly regulated system of amplification, and in hemophilia A or B, a critical amplifier is missing or deficient. The consequences, when left untreated or inadequately treated, are relentless: joint bleeds that destroy cartilage and deform limbs, muscle hemorrhages that compress nerves and blood vessels, and intracranial bleeding that can be fatal. For patients with a specific complication called inhibitors, even the standard replacement therapy stops working, leaving them in a position where a category of medications designed to save their lives has become ineffective against their own immune response.
Hympavzi (marstacimab-hncq), originally approved in October 2024, takes an entirely different approach to hemophilia prophylaxis. Rather than replacing the deficient clotting factor, it blocks a natural brake on the coagulation system: tissue factor pathway inhibitor (TFPI). On June 8, 2026, the FDA expanded Hympavzi’s approved indication to two of the groups most in need of this approach: patients with hemophilia A or B who have inhibitors (where factor replacement therapy either cannot work or works inadequately), and children aged 6 to 11 years, the youngest pediatric age group that can benefit from a once-weekly subcutaneous prophylactic therapy.
This post covers what hemophilia is, why inhibitors represent such a significant clinical challenge, how the TFPI mechanism works and why it is effective across both hemophilia types and inhibitor-positive patients, what the BASIS and BASIS KIDS trial data shows, and what the expanded approval means for families and clinicians managing these patients.
What Hemophilia Is: A Brief Overview
Hemophilia is a rare, X-linked recessive bleeding disorder caused by deficiency or dysfunction of specific coagulation factors. Two major types are recognized:
Hemophilia A is caused by deficiency or dysfunction of factor VIII (FVIII). It is the more common type, affecting approximately 1 in 5,000 male births.
Hemophilia B is caused by deficiency or dysfunction of factor IX (FIX). It affects approximately 1 in 25,000 male births.
Both conditions result in a dysfunctional coagulation cascade, the sequential activation of clotting proteins that ultimately converts fibrinogen to fibrin and forms a stable blood clot. Without adequate FVIII or FIX activity, the intrinsic pathway of coagulation cannot amplify adequately, resulting in delayed, insufficient clot formation. Minor injuries that healthy individuals handle without consequence become prolonged bleeding events in hemophilia. Spontaneous joint and muscle bleeds, which occur without obvious trauma, are a hallmark of severe hemophilia and are responsible for the progressive arthropathy (joint destruction) that historically defined the long-term morbidity of the disease.
The standard of care for moderate to severe hemophilia has been prophylactic factor replacement: infusing FVIII or FIX concentrate on a scheduled basis, typically two to three times per week for IV formulations or less frequently for extended half-life products, to maintain factor levels high enough to prevent spontaneous bleeding. This approach has dramatically improved outcomes over the past four decades. It requires, however, venous access and intravenous administration, which is particularly challenging in young children whose veins are small and who may require port placement.
The inhibitor problem
Inhibitors are neutralizing antibodies against the infused clotting factor, most commonly FVIII in hemophilia A. They develop in approximately 30% of patients with severe hemophilia A and in a smaller proportion of hemophilia B patients, typically early in treatment. When inhibitors are present, the infused factor is rapidly neutralized before it can participate in clotting, and standard prophylaxis becomes ineffective.
Managing hemophilia with inhibitors has historically required bypassing agents, products that activate the coagulation cascade at steps downstream of where FVIII and FIX normally act: recombinant factor VIIa (NovoSeven) and activated prothrombin complex concentrate (aPCC, FEIBA). These agents work differently from standard factor replacement and are less precisely dosed, making achieving predictable bleeding protection more challenging. Their use is also expensive, requires intravenous administration, and when used on-demand (only when bleeding occurs) rather than prophylactically, leaves patients with significant residual bleeding burden.
| Why non-factor therapies are a breakthrough for inhibitor patients The development of non-factor hemostatic therapies, which restore hemostasis by mechanisms that bypass the deficient factor entirely, represents one of the most significant advances in hemophilia management in decades. Emicizumab (Hemlibra), approved in 2017 for hemophilia A with inhibitors, was the first such therapy and demonstrated dramatic reductions in bleeding rates in a population that had previously had very limited prophylactic options. It works by bridging FIXa and FX, mimicking the co-factor function of FVIII specifically. However, because emicizumab bridges FIXa (factor IX in activated form), it works only in hemophilia A: it provides no benefit in hemophilia B because FIX is not a bridging partner for its mechanism. Marstacimab’s TFPI inhibition mechanism is active in both hemophilia A and hemophilia B regardless of inhibitor status, because it restores hemostasis upstream of where either FVIII or FIX deficiency disrupts the cascade. This is why Hympavzi’s expanded indication covers both hemophilia types and both inhibitor-positive and inhibitor-negative patients. |
|---|
The Science: How Marstacimab Works
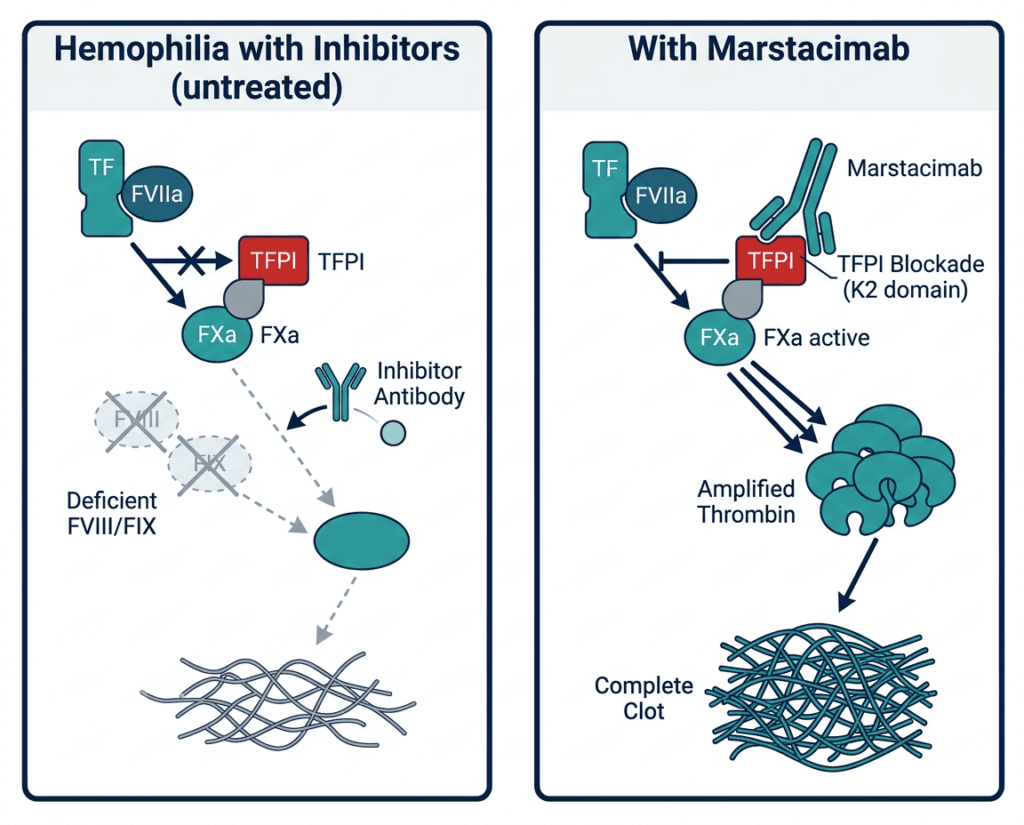
Tissue factor pathway inhibitor (TFPI) is a naturally occurring anticoagulant protein. It functions as a feedback inhibitor of the extrinsic pathway of coagulation: after tissue factor and factor VIIa initiate clotting in response to vascular injury, TFPI rapidly inhibits the TF-FVIIa-FXa complex, effectively braking the initial clotting signal after it has had time to initiate.
In healthy individuals, this brake is appropriate. The extrinsic pathway generates only a small initial burst of thrombin, and the intrinsic pathway (which requires FVIII and FIX) amplifies this initial signal to generate the large amounts of thrombin needed for stable clot formation. When FVIII or FIX is deficient, the initial thrombin burst generated by the extrinsic pathway is inadequate, and the amplification step through the intrinsic pathway cannot compensate. TFPI’s braking of the extrinsic pathway further limits the already insufficient hemostatic response.
Marstacimab is a human monoclonal antibody that targets the Kunitz 2 (K2) domain of TFPI with high specificity. The K2 domain is the site through which TFPI inhibits FXa, a critical downstream clotting protein. By blocking the K2 domain, marstacimab prevents TFPI from inhibiting FXa as efficiently. This allows the coagulation cascade to generate more thrombin from the extrinsic pathway initiation step, partially compensating for the deficient intrinsic amplification. The result is a shift in the coagulation balance toward clot formation even in the absence of adequate FVIII or FIX.
Critically, this mechanism functions regardless of:
- Whether the patient is deficient in FVIII (hemophilia A) or FIX (hemophilia B)
- Whether inhibitory antibodies are present against FVIII or FIX
- The severity of the underlying factor deficiency
Because marstacimab acts on a checkpoint in the coagulation cascade that is common to both hemophilia types and is entirely upstream of where inhibitors act against replacement factors, it provides hemostatic coverage across the full breadth of the expanded indication.
The Original Approval and What the Expansion Adds
Original approval (October 2024)
Hympavzi was initially approved in October 2024 for routine prophylaxis in patients aged 12 years and older with hemophilia A without FVIII inhibitors or hemophilia B without FIX inhibitors, based on the Phase 3 BASIS trial in the non-inhibitor cohort.
The June 2026 expansion
The June 8, 2026 expansion adds two critically important populations:
Population 1: Patients aged 12 years and older with hemophilia A or B who have inhibitors. This is the group with the most limited treatment options prior to this approval, for whom on-demand bypassing agents were often the only recourse for bleeding management.
Population 2: Pediatric patients aged 6 to 11 years with hemophilia A or B, with or without inhibitors. This extends Hympavzi’s reach to younger children, a population in whom venous access for IV therapy is a significant practical challenge, and for whom a once-weekly subcutaneous injection is potentially far more manageable than frequent IV infusions.
Together, the expanded and original indications give Hympavzi a single consolidated label: routine prophylaxis for adults and pediatric patients aged 6 years and older with hemophilia A or B, with or without inhibitors.
The Clinical Evidence: BASIS Inhibitor Cohort and BASIS KIDS
BASIS trial (NCT03938792): the inhibitor cohort data
BASIS (NCT03938792) was a Phase 3, global, open-label trial evaluating marstacimab in patients with hemophilia A or B with and without inhibitors. The trial included an inhibitor cohort of patients who had previously received on-demand (OD) bypassing agent therapy for bleeding episodes. This population represents the clinical severity of inhibitor disease: patients who had not been on prophylaxis and were relying on reactive treatment when bleeds occurred.
The primary efficacy comparison in the inhibitor cohort was marstacimab prophylaxis versus on-demand bypassing agent therapy. The results were striking:
| Outcome | Marstacimab prophylaxis | On-demand bypassing agent (historical) | Comparison |
|---|---|---|---|
| Mean treated annualized bleeding rate (ABR) | 1.4 (95% CI 0.9 to 2.3) | 19.8 (95% CI 16.1 to 24.3) | 93% reduction; ratio 0.07 (95% CI 0.04 to 0.12); p less than 0.0001 |
Source: Clinical Advisor. Hympavzi Gains Expanded FDA Approval for Hemophilia A and B. June 2026. Hematology Advisor. June 2026. Pfizer press release, June 8, 2026.
A 93% reduction in mean treated annualized bleeding rate compared to on-demand bypassing therapy is a clinically substantial finding. Patients going from a mean treated ABR of approximately 19.8 bleeds per year to 1.4 bleeds per year is a transformation in disease burden. The most devastating consequence of hemophilia with inhibitors has always been the inability to prevent spontaneous joint bleeds because on-demand therapy, by definition, does not begin until bleeding has already started. Each prevented bleed under prophylaxis is a joint or muscle or organ that did not bleed.
This comparison is against on-demand therapy because patients in the inhibitor cohort had not previously been on prophylaxis. An interpretive note: comparing against on-demand is a favorable comparison for the active treatment; a head-to-head comparison against existing prophylactic bypassing agent regimens would be more demanding. Nonetheless, the magnitude of the reduction is substantial and clinically meaningful.
BASIS KIDS trial (NCT05611801): the pediatric data
BASIS KIDS (NCT05611801) was a Phase 3, global, open-label trial evaluating the safety and efficacy of marstacimab in children aged 1 to 17 years with hemophilia A or B, with or without inhibitors. Interim results from this trial supported the FDA’s expanded approval for the 6-to-11-year age group.
| Population | Mean ABR with marstacimab | Comparison (historical model-based) |
|---|---|---|
| Ages 6 to 17, all patients (overall BASIS KIDS) | 1.8 | 3.6 (prior routine prophylaxis) |
| Ages 6 to under 18 with inhibitors (n=14) | 1.4 (99% CI 0.5 to 4.5) | 18.9 (99% CI 14.2 to 25.2) |
| Ages 6 to 11 cohort (n=7) | 1.3 (99% CI 0.5 to 3.4) | — |
Source: Clinical Advisor. CheckRare. BASIS KIDS NCT05611801.
The pediatric inhibitor subgroup data is particularly notable: children with hemophilia and inhibitors achieved a mean ABR of 1.4 with marstacimab prophylaxis compared to a historical model-based mean of 18.9 with on-demand bypassing therapy. For the youngest cohort (ages 6 to 11), the mean ABR of 1.3 indicates that the same degree of bleeding rate reduction is achievable in younger children.
The comparison in BASIS KIDS against historical model-based rates (rather than a randomized parallel arm) is appropriate for a pediatric rare disease trial, where randomizing young children to a control arm receiving inferior therapy would be ethically difficult and practically challenging. The historical modeling approach uses well-characterized natural history data from the hemophilia population to provide a reference benchmark.
What Makes This Approval Particularly Significant
The hemophilia B inhibitor gap
The most significant first-in-class designation from this expansion is that Hympavzi is now the first subcutaneous non-factor therapy available for children aged 6 to 11 years with hemophilia B. This deserves specific explanation.
Emicizumab (Hemlibra), the landmark non-factor therapy approved for hemophilia A with inhibitors in 2017, works by bridging activated factor IXa and factor X, mimicking the cofactor function of FVIII. This mechanism is specific to hemophilia A: it does not provide any benefit in hemophilia B because factor IXa is not the relevant enzyme that needs a cofactor. Patients with hemophilia B and inhibitors have therefore had no approved subcutaneous prophylactic non-factor therapy option. For them, marstacimab’s TFPI inhibition mechanism, which is indifferent to whether the underlying deficiency is in FVIII or FIX, represents the first approval in this category.
Once-weekly subcutaneous administration without routine laboratory monitoring
Hympavzi is administered once weekly by subcutaneous injection. The therapy does not require routine treatment-related laboratory monitoring, which distinguishes it from many hemophilia treatments that require regular factor level measurement or trough level testing to guide dosing. For families managing pediatric hemophilia, reducing the blood draw burden alongside simplifying the injection schedule to once weekly is a meaningful quality-of-life improvement.
Patients aged 12 years and older may self-inject after proper training. For children aged 6 to 11, caregiver administration is expected.
Safety: What the Prescribing Information Covers
The prescribing information for Hympavzi includes several important warnings:
Thromboembolic events: Two thromboembolic events occurred among 259 patients in the open-label extension study. Because marstacimab shifts the coagulation balance toward clot formation by removing the TFPI brake on the extrinsic pathway, the theoretical risk of thrombosis is inherent to its mechanism of action. Monitor patients for signs and symptoms of thromboembolic events. The absolute rate was low (2 of 259 patients in the extension), but the mechanism-based concern means clinical vigilance is appropriate.
Laboratory changes: Increases in fibrin D-dimer and prothrombin fragment 1.2 have been observed with marstacimab. These are laboratory markers of coagulation activation and fibrin turnover. They are consistent with the drug’s mechanism (enhanced coagulation pathway activity) but require awareness in clinical monitoring. Do not interpret elevated D-dimer as diagnostic for thrombosis without clinical correlation in patients receiving marstacimab.
Hypersensitivity reactions: As with all monoclonal antibodies, hypersensitivity reactions including anaphylaxis may occur. Patients should be informed of the symptoms and instructed to seek immediate medical attention if they occur.
Embryo-fetal toxicity: Based on the mechanism of action (promoting coagulation), there is a potential for fetal harm. Females of reproductive potential should use effective contraception during treatment.
Common adverse reactions (2% or greater): Injection site reactions, headache, pyrexia, arthralgia, diarrhea, pruritus, and rash.
What This Means for Patients and Families
For patients with hemophilia A or B who have inhibitors
This is the population for whom this expansion matters most urgently. Inhibitor-positive patients, particularly those with hemophilia B inhibitors, have had no approved subcutaneous non-factor prophylactic therapy before this date. The BASIS inhibitor cohort’s 93% reduction in treated ABR compared to on-demand bypassing therapy represents a shift from a reactive management strategy to an active preventive one, with the bleed rate reduction that entails.
If you or a family member has hemophilia with inhibitors and has been managed with on-demand bypassing agents, a conversation with your hematologist about whether Hympavzi is an appropriate prophylactic option is warranted.
For families of children aged 6 to 11
For young children with hemophilia, IV access for factor infusions has historically been the most practically difficult aspect of management, often requiring port placement and the risks that entails. A once-weekly subcutaneous injection administered by a caregiver at home, without routine blood draws for monitoring, represents a materially simpler treatment paradigm for this age group. The BASIS KIDS data showing a mean ABR of 1.3 in the 6-to-11 cohort supports the clinical adequacy of this approach.
For children with hemophilia B specifically, this is the first approved subcutaneous non-factor prophylactic therapy in this age group, filling a gap that emicizumab’s mechanism could not address.
For clinicians managing hemophilia
The complete current Hympavzi indication covers hemophilia A and B, with or without inhibitors, in patients aged 6 years and older. This is a single once-weekly subcutaneous platform applicable across the majority of the hemophilia population. For patients who are appropriate candidates, the elimination of routine treatment-related laboratory monitoring removes a significant ongoing management burden.
The thromboembolic warning and the D-dimer/prothrombin fragment 1.2 elevations associated with marstacimab require clinical awareness, particularly in patients who may develop thrombotic risk factors over time (surgery, immobilization, pregnancy). Risk-benefit discussions individualized to each patient’s circumstances remain the appropriate framework.
For related HED coverage on other rare hematologic disease approvals in 2026, see our post on KRESLADI, the first gene therapy approved for severe leukocyte adhesion deficiency type I and our post on Decnupaz (pivekimab sunirine) approved for blastic plasmacytoid dendritic cell neoplasm.
Sources
Pfizer FDA approval press release: U.S. FDA Approves Pfizer’s HYMPAVZI for the Treatment of Two Additional Hemophilia A or B Patient Populations with Significant Medical Need. Pfizer. June 8, 2026.
Drugs.com approval news: U.S. FDA Approves Pfizer’s Hympavzi for the Treatment of Two Additional Hemophilia A or B Patient Populations with Significant Medical Need. drugs.com. June 8, 2026.
BioPharm International clinical coverage: FDA Expands Pfizer’s Hympavzi Approval to Pediatric Hemophilia Patients and Those with Inhibitors. biopharminternational.com. June 2026.
Hematology Advisor (BASIS inhibitor data): Hympavzi Gains Expanded FDA Approval for Hemophilia A and B. hematologyadvisor.com. June 2026.
Clinical Advisor detailed summary: Hympavzi Gains Expanded FDA Approval for Hemophilia A and B. clinicaladvisor.com. June 2026.
Conexiant clinical summary: FDA Expands Marstacimab Indication in Hemophilia A and B. conexiant.com. June 2026.
CheckRare clinical coverage: FDA Expands Approval of Hympavzi (Marstacimab) for Patients With Hemophilia. checkrare.com. June 2026.
BASIS trial registration: NCT03938792. ClinicalTrials.gov.
BASIS KIDS trial registration: NCT05611801. ClinicalTrials.gov.
Priority Review grant (sBLA): FDA Grants Priority Review for HYMPAVZI sBLA. Pfizer. 2026.
Hympavzi original approval (October 2024): Hympavzi FDA Approval History. drugs.com.
Hympavzi prescribing information: HYMPAVZI (marstacimab-hncq) Prescribing Information. Pfizer. 2026.
TFPI biology and marstacimab mechanism: Tissue Factor Pathway Inhibitor. PMC6126283.
Coagulation cascade: Coagulation Studies. StatPearls. NCBI.
Hemophilia A overview: Hemophilia A. StatPearls. NCBI.
Hemophilia B overview: Hemophilia B. StatPearls. NCBI.
Hemophilia inhibitors: Factor VIII Inhibitors in Hemophilia A. PMC7155173.
CDC hemophilia overview: Hemophilia. CDC.
Patient resources: National Hemophilia Foundation | World Federation of Hemophilia | Pfizer Hympavzi patient support
| Disclaimer: Health Evidence Digest provides general information about FDA approvals and health research for educational purposes. This content is not a substitute for professional medical advice. Hemophilia management is complex and requires individualized assessment by a hematologist experienced in bleeding disorders. Decisions about switching to or initiating Hympavzi should be made in consultation with a treating hematologist who can assess the patient’s inhibitor status, bleeding history, current prophylaxis regimen, and individual risk-benefit profile. |
|---|
Leave a Reply