| 📌 The essentials On May 13, 2026, the FDA approved Inqovi (decitabine and cedazuridine, Taiho Oncology) in combination with venetoclax for adults with newly diagnosed acute myeloid leukemia (AML) who are 75 years or older, or who have comorbidities that preclude intensive induction chemotherapy. This is Inqovi’s third FDA-approved indication, joining myelodysplastic syndromes (MDS) and chronic myelomonocytic leukemia (CMML), both approved in 2020. What makes it first-in-class: Inqovi plus venetoclax is the first and only all-oral hypomethylating agent (HMA) plus BCL-2 inhibitor combination approved in any cancer setting. The previous standard of care for this population, azacitidine plus venetoclax (Venclexta), requires intravenous or subcutaneous azacitidine injections. The clinical basis: Phase 2b ASCERTAIN-V trial (NCT04657081), 101 adults with newly diagnosed AML ineligible for intensive chemotherapy. CR rate: 46.5% (95% CI 36.5 to 56.7%). CR/CRi rate: 63.4%. Median time to CR: 2.4 months. 75.3% of CR patients maintained remission at 12 months. Median OS: 15.5 months (95% CI 7.6 to not estimable). Safety: Grade 3 or higher adverse events in 98% of patients, consistent with known HMA/BCL-2 inhibitor class profile; febrile neutropenia, anemia, and neutropenia most frequent; no new drug-drug interaction signals. Dosing: Inqovi (35 mg decitabine / 100 mg cedazuridine) orally on days 1 to 5 of each 28-day cycle, with venetoclax 400 mg daily after cycle 1 ramp-up. All oral. No clinic infusion required. |
|---|
Acute myeloid leukemia is among the most challenging blood cancers to treat. It is diagnosed in an estimated 22,720 Americans in 2026, and it has an uncomfortable demographic reality: it predominantly affects older adults, with a median diagnosis age of approximately 68 years. The same patients who are most likely to develop AML are often the least able to tolerate the treatment that offers the best chance of remission, intensive induction chemotherapy involving days of hospitalization, profound bone marrow suppression, and significant mortality risk even when the treatment works.
For older adults and those with significant comorbidities, the established approach over the past five years has been a less intensive regimen of azacitidine or decitabine, both given by injection or infusion, combined with venetoclax (Venclexta, AbbVie/Genentech), an oral BCL-2 inhibitor. This HMA/BCL-2 combination transformed the treatment landscape for “unfit” AML patients after the pivotal VIALE-A trial, establishing remission rates and survival outcomes that were meaningfully better than HMA alone.
The problem is the parenteral component. Azacitidine requires injections or infusions on 7 days of every 28-day cycle, meaning patients must come to a clinic or have a home health nurse administer the drug regularly throughout treatment. For a 78-year-old with heart failure, limited mobility, or inadequate transport, this creates a real and sometimes insurmountable access barrier.
On May 13, 2026, the FDA approved a solution to that access problem: Inqovi (decitabine and cedazuridine) plus venetoclax, an all-oral alternative that achieves equivalent drug exposure to intravenous decitabine through a pharmacological workaround, administered entirely at home.
What Inqovi Is and the Problem It Was Designed to Solve
Inqovi is a fixed-dose combination tablet containing two drugs: decitabine and cedazuridine. Understanding why both are needed together requires understanding why oral decitabine alone does not work.
Decitabine: the hypomethylating agent
Decitabine is a nucleoside analog that works as a DNA hypomethylating agent (HMA). In cancer cells, abnormal DNA methylation silences tumor suppressor genes that would otherwise limit malignant growth. Decitabine incorporates into DNA and inhibits DNA methyltransferase (DNMT), reversing this silencing and restoring normal gene expression patterns. It is a well-established and effective anti-leukemia mechanism.
The problem is delivery. Decitabine taken orally is rapidly broken down in the gut and liver by an enzyme called cytidine deaminase (CDA) before it can reach the bloodstream in therapeutic concentrations. This is why intravenous decitabine and subcutaneous azacitidine have been used for decades: bypassing the gastrointestinal route avoids this degradation.
Cedazuridine: the CDA inhibitor that makes oral delivery work
Cedazuridine is a selective inhibitor of cytidine deaminase in the gut and liver. When cedazuridine is co-administered with decitabine, it blocks the enzyme responsible for degrading decitabine in the gastrointestinal tract, allowing therapeutic concentrations of decitabine to be absorbed and to reach systemic circulation.
Pharmacokinetic studies comparing oral Inqovi to IV decitabine 20 mg/m² demonstrated bioequivalent systemic exposure. The AUC (area under the concentration-time curve) for decitabine given as Inqovi matches IV decitabine, which was the key regulatory and clinical evidence establishing that the pill delivers the same drug at the same concentration as the infusion. This equivalence was demonstrated in the pivotal ASCERTAIN trial that supported Inqovi’s original 2020 approval for MDS and CMML.
Venetoclax: the BCL-2 inhibitor that completes the combination
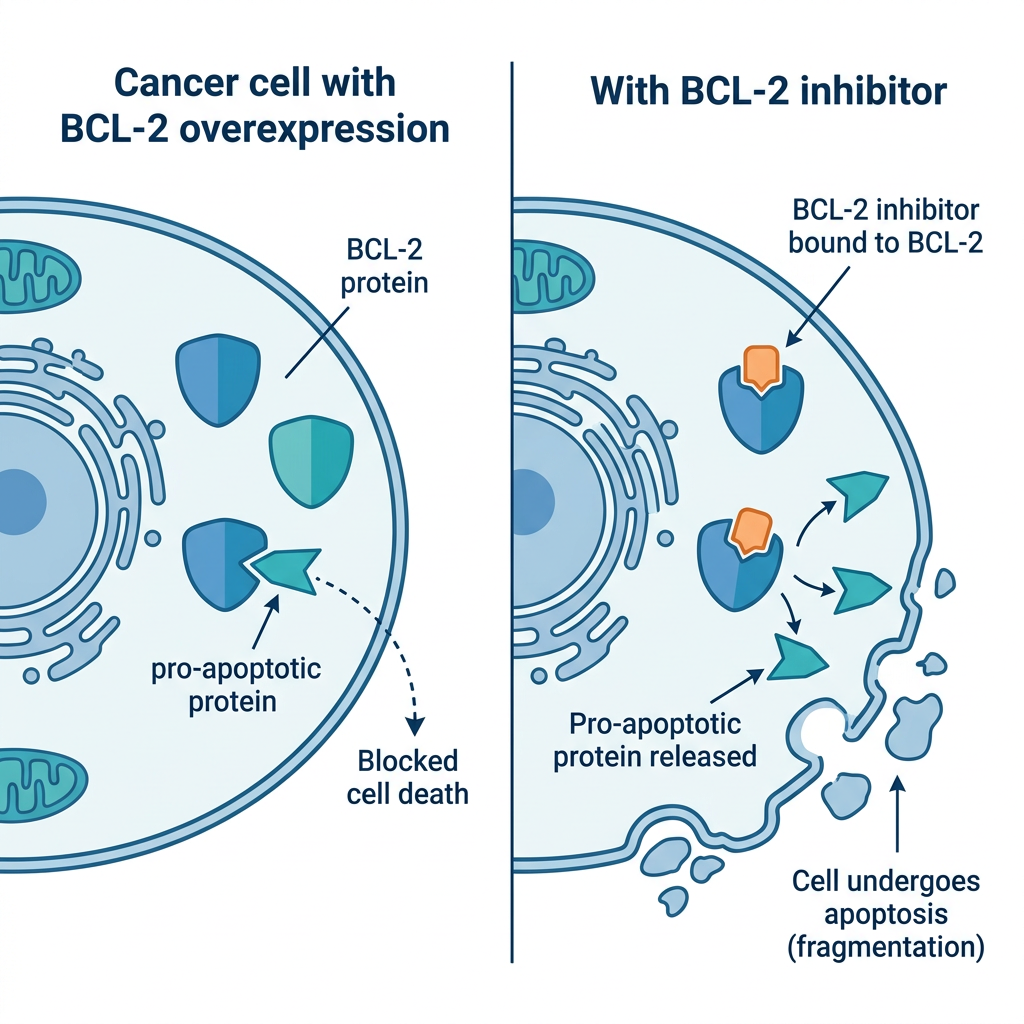
Venetoclax (Venclexta) is an oral BCL-2 inhibitor that works through a completely distinct mechanism: it blocks the BCL-2 protein, which cancer cells overexpress to prevent their own programmed death (apoptosis). By blocking BCL-2, venetoclax restores the apoptotic pathway, causing AML cells to die. The synergy between HMA and BCL-2 inhibition is well-established: HMA therapy primes AML cells toward apoptosis, and venetoclax completes the process.
Venetoclax has been oral since its approval. The barrier to an all-oral HMA/BCL-2 regimen was always the parenteral HMA component. Inqovi resolves that barrier.
| Why “all-oral” matters clinically beyond convenience The shift from intravenous azacitidine to oral Inqovi is not merely a quality-of-life upgrade, though quality of life matters significantly in older patients with AML. It has tangible clinical implications. Parenteral HMA regimens require 7 treatment days per cycle for azacitidine, meaning patients must arrange transportation to a clinic or home health services 7 times per month, often indefinitely. For a patient who lives alone, relies on others for transportation, or has mobility limitations from heart failure, pulmonary disease, or peripheral neuropathy, these logistical demands create real discontinuation risk. Studies in AML and MDS have documented that patients on parenteral HMA regimens miss doses or discontinue treatment earlier than protocol intent due to the burden of frequent clinic visits. An oral regimen administered at home on days 1 to 5 of a 28-day cycle eliminates these specific barriers entirely. |
|---|
AML in Patients Ineligible for Intensive Chemotherapy: Who Is This Population?
The FDA-approved indication covers adults aged 75 or older, or adults with comorbidities that preclude intensive induction chemotherapy. This population has specific characteristics that make treatment selection different from younger, fit patients.
Intensive induction chemotherapy for AML, typically the “7+3” regimen of cytarabine and anthracycline, is effective but produces profound myelosuppression (bone marrow suppression) and requires weeks of inpatient hospitalization. Early mortality rates with intensive therapy approach 10 to 15% in older patients, largely from infections and organ toxicity during the aplastic period. For a patient aged 75 with renal insufficiency, uncontrolled heart failure, or severe pulmonary disease, the risk of dying from the treatment may exceed the benefit.
The ASCERTAIN-V trial population reflects this clinical reality: median age was 78 years, 51.5% had ECOG performance status of 1, and 16.8% had TP53 mutations (associated with particularly poor prognosis). The distribution across European LeukemiaNet (ELN) risk categories was: favorable 31.7%, intermediate 33.7%, adverse 29.7%, reflecting a realistic mix of disease risk rather than a cherry-picked low-risk population.
The ASCERTAIN-V Trial: Full Results
Design
ASCERTAIN-V (NCT04657081) is an international, multicenter, open-label, single-arm, Phase 2 study. The Phase 2b portion enrolled 101 adults with newly diagnosed AML per WHO 2016 criteria, ECOG performance status 0 to 3, ineligible for intensive induction chemotherapy, with a life expectancy of at least 3 months.
Dosing schedule per 28-day cycle:
- Cycle 1 (venetoclax ramp-up): venetoclax 100 mg day 1, 200 mg day 2, 400 mg day 3 through day 28; Inqovi days 1 to 5
- Cycle 2 onward: Inqovi days 1 to 5; venetoclax 400 mg daily
Median follow-up at time of analysis: 11.2 months.
Efficacy results
| Outcome | Result |
|---|---|
| Complete remission (CR) rate | 46.5% (95% CI 36.5 to 56.7%) |
| CR plus CRi (incomplete hematologic recovery) rate | 63.4% (95% CI 53.2 to 72.7%) |
| CR plus CRh (partial hematologic recovery) rate | 51.5% (95% CI 41.3 to 61.6%) |
| Median time to CR | 2.4 months |
| Patients in CR at 6 months | 80.0% |
| Patients in CR at 12 months | 75.3% |
| Median duration of CR | Not reached at time of analysis |
| Median overall survival | 15.5 months (95% CI 7.6 to not estimable) |
Source: ASCERTAIN-V Phase 2b results. Presented at ASCO 2025 Annual Meeting and EHA Congress 2025. Taiho Oncology press release, May 13, 2026. NCT04657081.
Contextualization against the standard of care
The obvious benchmark is the VIALE-A trial, which established azacitidine plus venetoclax as the standard for unfit AML. VIALE-A produced a CR rate of 24.2% and a median OS of 14.7 months in the venetoclax arm. The ASCERTAIN-V CR rate of 46.5% and median OS of 15.5 months are directionally favorable, though cross-trial comparison must be interpreted with caution given differences in patient selection, trial design, and follow-up periods.
| Why cross-trial comparisons must be interpreted cautiously ASCERTAIN-V and VIALE-A enrolled patients using different criteria, at different time periods, in different geographic mixes of sites. ASCERTAIN-V’s single-arm design without a comparator arm means there is no concurrent control to account for evolving supportive care standards, referral patterns, or patient selection over time. The CR rates may also reflect different definitions or measurement timing. The appropriate conclusion from ASCERTAIN-V is that the all-oral combination achieves remission and survival outcomes consistent with published HMA/venetoclax benchmarks in this population, not that it is proven superior to azacitidine/venetoclax. A randomized confirmatory trial comparing the two approaches directly would be needed to establish superiority. |
|---|
Safety results
Safety results from ASCERTAIN-V were consistent with the known profiles of both drugs in this class and in this patient population. These are not mild side effects: AML patients treated with HMA/BCL-2 inhibitor combinations experience significant myelosuppression, and the grade 3 or higher adverse event rate reflects this.
| Safety outcome | Rate |
|---|---|
| Grade 3 or higher adverse events (any) | 98.0% of patients |
| Febrile neutropenia | Most common grade 3 or higher event |
| Anemia (grade 3 or higher) | Frequent |
| Neutropenia (grade 3 or higher) | Frequent |
| Dose interruptions | 68.3% |
| Discontinuations due to adverse events | 8.9% |
| Fatal adverse events | 15.8% (3 attributed to adverse events, 10 to disease progression per 30/60-day mortality assessment) |
| New drug-drug interactions (venetoclax/decitabine) | None identified |
The 98% grade 3 or higher adverse event rate is alarming on first read but is consistent with what hematologists and patients should expect in AML treatment. Myelosuppression is inherent to HMA therapy in a disease characterized by dysfunctional bone marrow. The absence of new drug-drug interaction signals between oral decitabine and venetoclax (which was a theoretical concern given cedazuridine’s CDA inhibition affecting venetoclax metabolism) is an important safety finding.
Venetoclax requires co-administration with a CYP3A4 inhibitor prophylaxis protocol during ramp-up. Patients on antifungal prophylaxis (such as posaconazole or fluconazole) need venetoclax dose reductions during ramp-up because these agents inhibit CYP3A4 and increase venetoclax exposure. Prescribers managing this combination must follow the venetoclax drug interaction tables carefully.
Safety Warnings: What the Label Covers
The Inqovi prescribing information includes important safety warnings that apply to the AML indication:
Myelosuppression: Inqovi can cause severe myelosuppression in patients with AML, including fatal adverse reactions. Complete blood count monitoring before each treatment cycle and as clinically indicated is required.
Differentiation syndrome: HMA agents and venetoclax can cause differentiation syndrome in AML, a potentially life-threatening inflammatory reaction caused by rapid differentiation of leukemic cells. Symptoms include fever, dyspnea, hypoxemia, rapid weight gain, and pleural or pericardial effusion. This is a medical emergency requiring corticosteroids and sometimes treatment interruption.
Embryo-fetal toxicity: Inqovi can cause fetal harm. Females of reproductive potential should use effective contraception during treatment and for at least 6 months after the final dose. Males with female partners of reproductive potential should use effective contraception during treatment and for at least 3 months after the final dose. Breastfeeding should not occur during treatment and for at least 2 weeks after the final dose.
Interactions with venetoclax: Strong or moderate CYP3A4 inhibitors increase venetoclax exposure and require dose adjustments. Avoid strong CYP3A4 inducers. The full venetoclax prescribing information and dose modification tables for drug interactions apply throughout the regimen.
Dosing: The All-Oral Treatment Schedule
The ASCERTAIN-V-based treatment schedule is:
Cycle 1 (venetoclax ramp-up cycle):
- Venetoclax: 100 mg on day 1, 200 mg on day 2, 400 mg on days 3 through 28
- Inqovi (35 mg decitabine / 100 mg cedazuridine): one tablet orally once daily on days 1 to 5
- Antifungal prophylaxis should accompany the venetoclax ramp-up per institutional protocol and the venetoclax prescribing information
Cycles 2 and beyond (28-day cycles):
- Inqovi: one tablet orally once daily on days 1 to 5
- Venetoclax: 400 mg orally once daily on days 1 to 28 (or per dose modification per prescribing information)
Inqovi tablets should be taken at approximately the same time each day. They can be taken with or without food. The tablet should be swallowed whole; do not cut, crush, or chew. If a dose is missed, it should be taken as soon as possible on the same day. If the day has passed, the missed dose should be skipped and the next dose taken at the regular scheduled time.
Where This Approval Fits: Inqovi’s Full Indication Picture
With this approval, Inqovi now carries three FDA-approved indications across blood cancers:
| Indication | Date | Partner drug |
|---|---|---|
| Myelodysplastic syndromes (MDS), including CMML | July 2020 | None (monotherapy) |
| Newly diagnosed AML, intensive chemotherapy-ineligible | May 13, 2026 | Venetoclax (Venclexta) |
The progression from MDS/CMML monotherapy to AML combination therapy reflects the well-established clinical overlap between these diseases. Many patients with high-risk MDS progress to AML, and the HMA mechanism is active across the disease spectrum. The ability to use the same oral drug platform in both MDS and AML simplifies the treatment transition for patients who progress from one to the other.
For Patients and Caregivers
Who qualifies?
Adults newly diagnosed with AML who are either 75 years or older, or who have comorbidities (organ dysfunction, severe frailty, concurrent illness) that make intensive induction chemotherapy unsafe. This is not a designation based solely on age: younger patients with significant comorbidities may also qualify.
What does all-oral treatment mean practically?
The five-day Inqovi dosing window (days 1 to 5 of each cycle) combined with daily venetoclax means treatment is taken at home throughout each 28-day cycle. There is no scheduled infusion clinic requirement for the regimen itself. Blood count monitoring, toxicity assessment, and clinical follow-up still require clinic visits, but not for drug administration.
This is a genuinely meaningful practical change for patients who live alone, lack reliable transportation, or have mobility limitations. The drug is given at home, and patients control their own administration schedule within the 5-day window.
Questions to discuss with your hematologist
- Do my specific AML genetics (cytogenetics, molecular markers) suggest I am likely to respond to an HMA/BCL-2 approach?
- Should I be tested for TP53, FLT3, IDH1/2, or other mutations that might inform therapy selection?
- How will venetoclax drug interactions with my current medications be managed, particularly if I am on antifungal prophylaxis?
- What blood count monitoring schedule is appropriate for my regimen?
- What are the early signs of differentiation syndrome and when should I call the oncology team?
For patients and families navigating a new AML diagnosis, the Leukemia and Lymphoma Society provides patient-facing disease information, clinical trial databases, and financial assistance resources. AAMDSIF (Aplastic Anemia and MDS International Foundation) covers the full MDS and AML spectrum with educational materials specific to these diseases. The NCI AML information page provides current treatment guidance in patient-accessible language.
For related coverage on venetoclax and its expanding role in blood cancers, including the first all-oral AML regimen and the Immgolis biosimilar approvals relevant to RA and UC, see our venetoclax coverage on Health Evidence Digest.
Sources
FDA approval announcement: FDA approves oral combination of decitabine and cedazuridine tablets with venetoclax for newly diagnosed acute myeloid leukemia. FDA.gov. May 13, 2026.
Taiho Oncology press release: U.S. FDA Approves INQOVI in Combination with Venetoclax, the First All-Oral Combination Treatment for Patients with Acute Myeloid Leukemia Who Are Ineligible for Intensive Induction Chemotherapy. businesswire.com. May 13, 2026.
Cancer Therapy Advisor approval coverage: All-Oral Inqovi Regimen Approved for Newly Diagnosed AML. cancertherapyadvisor.com. May 2026.
AJMC approval coverage: FDA Approves First All-Oral Regimen for AML. ajmc.com. May 2026.
Targeted Oncology approval coverage: FDA Approves Decitabine/Cedazuridine Plus Venetoclax in Unfit AML. targetedonc.com. May 2026.
OncLive approval coverage: FDA Approves Decitabine/Cedazuridine Plus Venetoclax in Newly Diagnosed AML. onclive.com. May 2026.
CancerNetwork ASCO 2025 results: Oral Venetoclax Combo Elicits Responses, Survival in Newly Diagnosed AML. cancernetwork.com. 2025.
AJMC ASCO 2025 summary: All-Oral AML Therapy: Decitabine-Cedazuridine Plus Venetoclax. ajmc.com. July 2025.
ASCERTAIN-V trial registration: NCT04657081. ClinicalTrials.gov.
Inqovi original MDS/CMML FDA approval: FDA approves decitabine and cedazuridine for treatment of adults with myelodysplastic syndromes. FDA.gov. July 2020.
Venetoclax AML FDA approval: FDA approves venetoclax in combination for treatment of adults with newly diagnosed acute myeloid leukemia. FDA.gov.
Decitabine mechanism: Decitabine. StatPearls. NCBI.
DNA methylation and HMA therapy: DNA Hypomethylating Agents in Myeloid Malignancies. PMC4207474.
Cedazuridine mechanism: Cedazuridine pharmacology. PMC7965890.
BCL-2 and venetoclax: BCL-2 Inhibitors in Hematologic Malignancies. PMC8626879.
AML overview: Acute Myeloid Leukemia. American Cancer Society.
ASCO 2025 Annual Meeting: ASCO 2025 Annual Meeting. asco.org.
EHA Congress 2025: EHA Congress 2025. ehaweb.org.
Patient resources: Leukemia and Lymphoma Society | AAMDSIF | NCI AML Treatment (PDQ) | ClinicalTrials.gov: AML
| Disclaimer: Health Evidence Digest provides general information about FDA approvals and health research for educational purposes. This content is not a substitute for professional medical advice, diagnosis, or treatment. Treatment decisions for acute myeloid leukemia, including eligibility for intensive versus less intensive regimens, should be made in close consultation with a board-certified hematologist or hematologic oncologist familiar with the patient’s full disease characteristics, performance status, and comorbidity profile. |
|---|