| 📌 The essentials Simponi (golimumab, Janssen/Johnson and Johnson) is a fully human anti-TNF-alpha monoclonal antibody approved for four indications: moderately to severely active rheumatoid arthritis (RA) in combination with methotrexate; active psoriatic arthritis; active ankylosing spondylitis; and moderately to severely active ulcerative colitis (UC) in patients who have had an inadequate response to prior therapy. Simponi Aria is an IV formulation approved for RA only. Simponi generated $1.19 billion in U.S. sales in 2025. Clinical basis: the GO-series Phase 3 trials (GO-FORWARD for RA, GO-RAISE for AS, GO-REVEAL for PsA) and the PURSUIT program for UC. ACR20 response at Week 14 in GO-FORWARD: 55.1% with golimumab 50 mg plus methotrexate versus 28.4% with placebo plus methotrexate. Five-year persistence: 69.8% of patients on golimumab as first-line therapy remained on treatment at Year 5 across all three arthritis indications. Why this LOE is different from others in this series: two biosimilar candidates exist, but both face significant obstacles as of mid-2026. AVT05 (Alvotech/Teva) received a Complete Response Letter from the FDA in November 2025 for manufacturing deficiencies at Alvotech’s Reykjavik facility; resubmission is planned. Immgolis and Immgolis Intri (golimumab-sldi, Bio-Thera/Accord) received FDA approval on May 15, 2026, making them the first approved biosimilars to Simponi and Simponi Aria — but commercial launch is currently blocked by a preliminary injunction motion Janssen filed on May 6, 2026. A hearing is expected August to September 2026. The practical result: no golimumab biosimilar is commercially available in the U.S. as of mid-2026, and the timeline for market entry remains uncertain. |
|---|
| 📚 About this series: the 2026 Loss of Exclusivity Watch This is Post 5 of HED’s 2026 Loss of Exclusivity series, tracking the ten major drugs losing U.S. exclusivity this year. The full series covers: Xolair (omalizumab) • Pomalyst (pomalidomide) • Opsumit (macitentan) • Januvia/Janumet (sitagliptin) • Simponi (golimumab) • Mavenclad (cladribine) • Gattex (teduglutide) • Trintellix (vortioxetine) • Briviact (brivaracetam) • Xeljanz (tofacitinib). Each post follows the same format: what the drug is and how it works, what the clinical evidence shows, who uses it and why, and what the entrance of competition means for patients, prescribers, and the market. |
|---|
The TNF inhibitor story is one of the most consequential chapters in modern medicine. Before etanercept launched in 1998, rheumatoid arthritis was a disease that reliably destroyed joints, disabled hands, ended careers, and shortened lives. The treatment options were methotrexate, sulfasalazine, hydroxychloroquine, and corticosteroids, agents that helped many patients but left a substantial proportion with progressive, irreversible damage regardless. The biological agents that followed — infliximab, etanercept, adalimumab, then golimumab (aka Simponi) — did not just improve outcomes. For many patients, they changed the entire trajectory of what the disease would do to them.
Golimumab is a fully human TNF-alpha inhibitor approved for moderate-to-severe rheumatoid arthritis in combination with methotrexate, active psoriatic arthritis, active ankylosing spondylitis, and moderately to severely active ulcerative colitis in patients with an inadequate response to prior therapy. Simponi generated $1.19 billion in U.S. sales in 2025, making it one of the largest drugs in the TNF inhibitor class and one of the ten biggest LOE stories of 2026.
But unlike most drugs in this series, Simponi’s transition to biosimilar competition is not proceeding on a simple timeline. As of mid-2026, no golimumab biosimilar is commercially available in the U.S. The first approved biosimilars, Immgolis and Immgolis Intri, received FDA approval on May 15, 2026 but are currently blocked from launch by active patent litigation. A second candidate, AVT05, received a manufacturing-related Complete Response Letter in November 2025 and is awaiting resubmission. Our dedicated post on the Immgolis approval covers the litigation and access timeline in full detail.
This post covers what golimumab is and how it works, what the GO-series clinical trials showed across all four indications, what makes golimumab distinctive among TNF inhibitors, what the safety requirements mean in practice, and what the litigation-constrained biosimilar landscape means for patients and payers.
What Golimumab Treats: Four Indications, One Mechanism
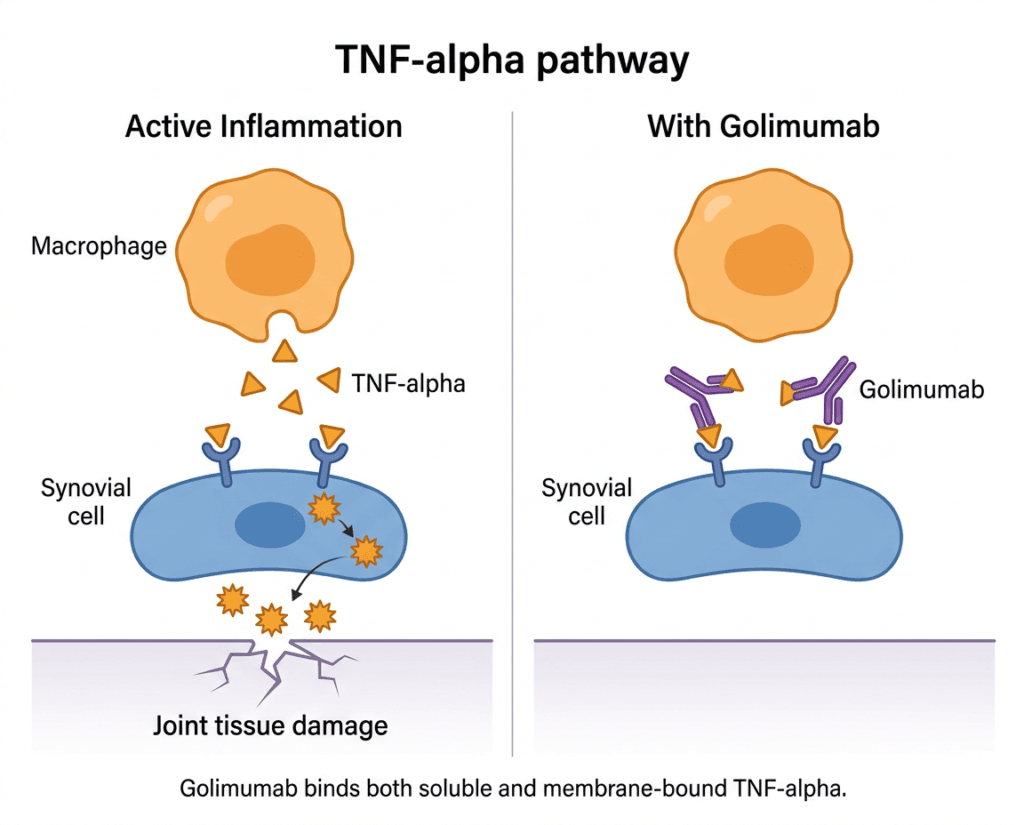
Tumor necrosis factor-alpha (TNF-alpha) is a pro-inflammatory cytokine whose overexpression is implicated in the pathophysiology of several chronic immune-mediated inflammatory diseases. Golimumab is a transgenic anti-TNF monoclonal antibody that binds both soluble and transmembrane forms of TNF-alpha, preventing binding to its receptors and inhibiting downstream inflammatory activity. Understanding each approved indication separately matters because the patients who use golimumab for RA are clinically, demographically, and therapeutically quite different from those who use it for ulcerative colitis, even though the drug’s mechanism is the same.
Rheumatoid arthritis (RA) is a chronic autoimmune disease in which the immune system attacks the synovial lining of joints, producing inflammation, pain, swelling, and, without adequate treatment, progressive joint destruction and disability. It affects roughly 1.5 million Americans, with a strong female predominance, and typically presents in middle age. TNF-alpha is one of the primary cytokines driving synovial inflammation in RA. Golimumab is approved for use with methotrexate in adults with moderate-to-severe active RA.
Psoriatic arthritis (PsA) is an inflammatory arthritis that occurs in approximately 30% of people with psoriasis. It has a heterogeneous clinical presentation: peripheral joint inflammation, axial disease, enthesitis, and dactylitis can all occur. TNF-alpha is elevated in psoriatic joint tissue, making TNF inhibition effective for both the skin and joint manifestations.
Ankylosing spondylitis (AS) and non-radiographic axial spondyloarthritis (nr-axSpA) are inflammatory conditions primarily affecting the spine and sacroiliac joints. Ankylosing spondylitis involves visible structural changes on imaging and can cause progressive spinal fusion; nr-axSpA involves active inflammatory disease without those radiographic changes. Both cause significant pain, stiffness, and functional impairment, and TNF-alpha is centrally involved in their pathogenesis. Golimumab is approved for both.
Ulcerative colitis (UC) is a chronic inflammatory bowel disease affecting the colon and rectum. TNF-alpha is a key driver of mucosal inflammation in UC, and golimumab was the first subcutaneous TNF inhibitor approved specifically for UC in 2013, based on the PURSUIT trial program.
The Science: What TNF-Alpha Is and Why Blocking It Works
TNF-alpha is a cytokine produced primarily by macrophages and T cells. In acute inflammation, it serves important functions: coordinating immune responses against infections, activating neutrophil killing of bacteria, and initiating fever as part of the body’s defensive response. In autoimmune disease, however, TNF-alpha production becomes chronically dysregulated.
In RA, the synovial tissue of affected joints is infiltrated by TNF-producing macrophages and activated T cells. The sustained high local concentrations of TNF-alpha drive ongoing inflammation, stimulate osteoclast-mediated bone erosion, and create a cycle of joint damage that continues even when the triggering event is long past. A similar dysregulated inflammatory loop operates in psoriatic arthritis, the spondyloarthropathies, and the bowel mucosa in ulcerative colitis.
Golimumab is a fully human monoclonal antibody that binds to both the soluble and transmembrane bioactive forms of human TNF-alpha, preventing TNF-alpha from binding to its receptors and thereby inhibiting its biological activity. This dual binding distinguishes golimumab from etanercept, which only binds soluble TNF. The clinical relevance of transmembrane TNF binding is most apparent in inflammatory bowel disease: etanercept has shown less efficacy than monoclonal anti-TNF antibodies in Crohn’s disease, thought to relate at least in part to transmembrane TNF signaling in granuloma formation.
Golimumab is a fully human antibody produced using transgenic mice with human antibody-producing genes, meaning the resulting antibody is entirely human in amino acid sequence. Infliximab is chimeric (part mouse, part human). Adalimumab is also fully human but was developed through phage display technology. Golimumab’s fully human structure theoretically reduces the risk of anti-drug antibody formation compared to chimeric antibodies, though immunogenicity in clinical practice varies across patients and is not reliably predicted by molecular origin alone.
Golimumab is available in two formulations: Simponi (subcutaneous injection, 50 mg every 4 weeks for most indications, delivered via prefilled syringe or SmartJect autoinjector) and Simponi Aria (intravenous infusion, 2 mg/kg at weeks 0 and 4, then every 8 weeks, approved for RA only). The subcutaneous formulation allows self-administration at home.
The GO-Series Clinical Trials: What the Evidence Shows
Golimumab’s clinical development program was named the GO-series: GO-FORWARD for RA, GO-RAISE for ankylosing spondylitis, GO-REVEAL for psoriatic arthritis, and PURSUIT for ulcerative colitis. Each was a Phase 3 randomized, double-blind, placebo-controlled study with methotrexate as background therapy where applicable.
Rheumatoid arthritis: GO-FORWARD
GO-FORWARD enrolled 444 patients with active RA despite stable methotrexate therapy, randomizing them to placebo plus methotrexate, golimumab 50 mg plus methotrexate, golimumab 100 mg plus methotrexate, or golimumab 100 mg alone. The primary endpoints were ACR20 response at Week 14 and HAQ-DI improvement at Week 24.
| Endpoint | Placebo plus MTX | Golimumab 50 mg plus MTX | Golimumab 100 mg plus MTX |
|---|---|---|---|
| ACR20 at Week 14 | 28.4% | 55.1% | 56.2% |
| ACR50 at Week 14 | 11.4% | 29.4% | 37.1% |
| ACR70 at Week 14 | 3.4% | 17.6% | 20.0% |
| HAQ-DI improvement 0.25 or greater at Week 24 | 29.5% | 56.9% | 57.8% |
Source: Keystone EC et al. Ann Rheum Dis. 2009;68(6):789–796. GO-FORWARD trial.
Clinical improvement was maintained through Week 104, with approximately 75% and 72% of patients randomized to golimumab 50 mg plus methotrexate and 100 mg plus methotrexate respectively achieving ACR20 response at two years. Radiographic data from GO-FORWARD also demonstrated that golimumab plus methotrexate inhibited structural damage progression compared to methotrexate alone, a finding reinforced in the GO-FURTHER IV study with Simponi Aria, where significant inhibition of radiographic progression was observed at weeks 24, 52, and 100.
Ankylosing spondylitis and psoriatic arthritis
GO-RAISE (ankylosing spondylitis) and GO-REVEAL (psoriatic arthritis) both demonstrated significant improvements in disease-specific outcome measures versus placebo, consistent with the established efficacy of TNF inhibitors in these conditions. A five-year pooled analysis of pivotal trial data including 2,228 patients with RA, psoriatic arthritis, and ankylosing spondylitis found golimumab retention rates at Year 5 were consistently high at 69.8% when used as first-line therapy, with no significant differences across the three indications. That five-year persistence figure is a meaningful real-world signal: patients who start golimumab tend to stay on it, suggesting sustained tolerability and ongoing benefit.
Ulcerative colitis: the PURSUIT trials
The PURSUIT program consisted of two trials: PURSUIT-SC (induction) and PURSUIT-Maintenance. In PURSUIT-SC, patients with moderate-to-severe UC despite conventional therapy were randomized to golimumab induction doses or placebo. Golimumab achieved significantly higher rates of clinical response and remission at Week 6, with response rates of approximately 55% for the 200/100 mg induction regimen versus 30% for placebo. In PURSUIT-Maintenance, patients who had achieved clinical response were randomized to golimumab 50 mg, golimumab 100 mg, or placebo every 4 weeks through Week 54. The 100 mg dose demonstrated maintenance of response significantly superior to placebo.
Golimumab’s approval for UC gave the gastroenterology community a subcutaneous option with a once-monthly home administration schedule. For patients who can self-inject, the convenience profile has been a meaningful factor in treatment choice compared to infliximab, which requires IV infusion at an infusion center.
How Golimumab Compares to Other TNF Inhibitors
Five TNF inhibitors are approved in the United States for inflammatory arthritis and related conditions: etanercept (Enbrel), infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), and golimumab (Simponi/Simponi Aria). No definitive head-to-head randomized controlled trials compare all five; rheumatologists select based on route of administration, dosing frequency, specific indication, patient preference, and payer formulary.
| Agent | Type | Route | Frequency | Half-life | Notable feature |
|---|---|---|---|---|---|
| Etanercept | Fusion protein (soluble TNF receptor) | SC | Weekly or biweekly | approximately 4 days | Only binds soluble TNF; less effective in IBD |
| Infliximab | Chimeric monoclonal antibody | IV infusion | Every 8 weeks after loading | approximately 9 to 12 days | First in class; broad indication history |
| Adalimumab | Fully human monoclonal antibody | SC | Every 2 weeks | approximately 14 days | Most prescribed biologic globally; extensive biosimilar competition now |
| Certolizumab pegol | PEGylated Fab fragment | SC | Every 2 or 4 weeks | approximately 14 days | No Fc region; may be preferred in pregnancy |
| Golimumab | Fully human monoclonal antibody | SC or IV | Monthly SC; every 8 weeks IV | approximately 12 to 14 days | Once-monthly SC dosing; only SC TNFi approved for UC |
The once-monthly subcutaneous dosing frequency of Simponi is a meaningful differentiator for patient experience. Compared to adalimumab’s biweekly injections or etanercept’s weekly or biweekly schedule, monthly injections reduce the injection burden substantially for patients managing chronic disease long-term.
The Safety Profile: What TNF Inhibition Means for Infection Risk
All TNF inhibitors carry a boxed warning for serious infections and malignancies, and golimumab is no exception. Understanding what this means in practice requires context. TNF-alpha is part of the immune system’s first-line defense against intracellular pathogens, particularly mycobacteria and certain fungal organisms. Blocking TNF-alpha reduces the immune system’s ability to contain latent infections, which is why TB reactivation is the most clinically important pre-treatment safety check for all TNF inhibitors.
In cases of reactivated latent tuberculosis, reactivation typically occurs within the first few months of treatment. Patients with latent tuberculosis should receive treatment with isoniazid or combination anti-tuberculosis agents before initiating any anti-TNF agent. Combining TNF-alpha inhibitor treatment with methotrexate or azathioprine further increases TB reactivation risk. Newer TNF inhibitors including golimumab have not been associated with a clearly increased TB risk compared to earlier agents adalimumab and infliximab, though ongoing surveillance continues.
| Safety item | Details | Clinical guidance |
|---|---|---|
| Serious infections (boxed warning) | Increased risk of bacterial, viral, fungal, and opportunistic infections, including fatal cases. Risk increases with concomitant immunosuppressives. | Evaluate for active infection before each dose. Hold golimumab if serious infection develops; do not resume until resolved. |
| Tuberculosis (boxed warning) | Risk of reactivation of latent TB, including disseminated or extrapulmonary cases. | Screen for latent TB with tuberculin skin test or IGRA before initiating. Treat latent TB before starting golimumab. Monitor during treatment. |
| Malignancy (boxed warning) | Lymphoma and other malignancies reported; hepatosplenic T-cell lymphoma cases reported primarily in adolescent and young adult males with IBD on concomitant immunosuppressives. | Discuss cancer risk with patients, particularly those with existing risk factors. Not recommended in patients with known malignancy other than treated skin cancer. |
| Hepatitis B reactivation | Reactivation of HBV in chronic carriers; some cases fatal. | Screen all patients for HBV before initiating. Monitor HBV carriers throughout treatment and after discontinuation. |
| Congestive heart failure | New onset or worsening; TNF inhibitors should not be used in patients with moderate-to-severe CHF (NYHA Class III/IV). | Avoid in moderate-to-severe CHF. Use with caution in mild CHF; monitor for worsening. |
| Demyelinating disease | Rare cases of new onset or exacerbation of demyelinating conditions including multiple sclerosis and Guillain-Barré syndrome. | Avoid in patients with known demyelinating disease. Consider discontinuing if neurological symptoms develop. |
| Drug-induced lupus | Anti-double-stranded DNA antibodies and drug-induced lupus reported; resolves on discontinuation. | Evaluate if lupus-like symptoms develop. |
| Live vaccines | Contraindicated during golimumab treatment. | Update all vaccinations before initiating therapy. No live vaccines during treatment. |
| Injection site reactions | Common with SC formulation: redness, bruising, pain at injection site. | Typically mild; rotate injection sites. |
The infection risk, while real, should be understood quantitatively. In pivotal trials, serious infections occurred at rates of approximately 2 to 6 per 100 patient-years in golimumab arms versus roughly 2 to 3 per 100 patient-years in placebo arms. The absolute individual patient risk in any given year is low, and the clinical benefit in controlling active inflammatory disease is substantial. The risk-benefit calculus is the domain of the prescribing rheumatologist or gastroenterologist who knows the patient’s full clinical picture.
The Biosimilar Landscape: Approved But Not Yet Available
This is where Simponi diverges sharply from most other drugs in this LOE series, and from the broader pattern of biologic LOEs like adalimumab (Humira), which saw dozens of biosimilars enter the U.S. market following its 2023 LOE.
As of mid-2026, no golimumab biosimilar is commercially available in the United States. Two candidates exist, and both have been significantly delayed.
AVT05 (Alvotech/Teva): AVT05 received its first global approval in Japan in September 2025 and a positive CHMP opinion in Europe the same month, where it is commercialized as Gobivaz. Alvotech’s BLA was accepted by the FDA in January 2025. On November 2, 2025, the FDA issued a Complete Response Letter for AVT05 citing manufacturing deficiencies identified during a pre-license inspection of Alvotech’s Reykjavik facility in July 2025. No other deficiencies were identified with the application. Alvotech has stated it expects to resolve the outstanding manufacturing issues and continues to work with the FDA toward bringing the biosimilar to U.S. patients.
Immgolis and Immgolis Intri (golimumab-sldi, Bio-Thera/Accord): The FDA accepted Bio-Thera and Accord’s abbreviated BLA for BAT2506 in July 2025. On May 15, 2026, the FDA approved Immgolis (golimumab-sldi) and Immgolis Intri (golimumab-sldi) as the first-ever interchangeable biosimilars to Simponi and Simponi Aria respectively. However, on May 6, 2026, Janssen had filed a BPCIA complaint against Accord and Bio-Thera in the U.S. District Court for the District of Delaware, identifying 17 patents, and a preliminary injunction motion to block launch followed. A hearing is expected August to September 2026. Accord BioPharma’s planned launch target remains Q4 2026, contingent on the litigation outcome.
The 17-patent listing signals an aggressive IP protection strategy. J&J has historically sought new patents for formulations, manufacturing methods, and treatment methods to extend market exclusivity beyond the initial core patent expiry. This approach has successfully delayed competitive entry across multiple biologic franchises.
The combined effect of the Alvotech manufacturing CRL and the Janssen-Bio-Thera preliminary injunction is that Simponi will almost certainly enter 2027 with no biosimilar competitor commercially available in the U.S. That is a materially different outcome from what happened with adalimumab, where 37 biosimilar versions received FDA approval after its 2023 LOE, creating intense competitive pressure and dramatic price reductions for payers.
For patients: the absence of a commercially available biosimilar does not change anything about Simponi’s availability or your current treatment. It does mean that the cost relief biosimilar competition typically delivers is delayed, possibly by a year or more.
For a detailed breakdown of the Immgolis/Immgolis Intri FDA approval, the indication scope differences from Simponi’s full label, the interchangeability designation, and the full litigation timeline, see our dedicated post: The First Biosimilars to Simponi and Simponi Aria Just Got FDA Approval. Here Is What Immgolis and Immgolis Intri Are, What They Treat, and Why You Cannot Buy Them Yet.
What This Means in the Broader TNF Inhibitor Market
The TNF inhibitor class is experiencing a profound market transition that predates Simponi’s LOE and will continue long after its biosimilars eventually launch. Adalimumab’s LOE in 2023 has dramatically reshaped biosimilar economics. With over 30 biosimilars approved and competition fierce, list prices for adalimumab products in the U.S. have dropped substantially. That competitive pressure has forced payers to renegotiate the entire TNF inhibitor category, including drugs that have not yet lost exclusivity. Simponi’s net price to many payers has almost certainly been reduced relative to its list price as payers leverage the adalimumab biosimilar market to extract rebates from all originator biologics.
For patients on golimumab who are well-controlled: this existing market pressure means J&J has ongoing incentive to keep Simponi competitively priced relative to adalimumab biosimilars. The delay in Simponi biosimilar entry does not mean payers are simply paying full list price. Formulary negotiations and rebate structures are continuously active even without a direct biosimilar competitor.
For patients newly initiating TNF inhibitor therapy for RA, PsA, or AS: adalimumab biosimilars are now among the lower-cost biologic options and are increasingly preferred by many formularies. Whether golimumab offers a clinical advantage sufficient to justify a premium over adalimumab biosimilars is a question for your rheumatologist, who will weigh dosing frequency preferences, prior treatment history, and individual patient factors.
For patients with ulcerative colitis: golimumab’s subcutaneous UC indication sets it apart. Adalimumab also has a UC indication, but infliximab IV remains the most established anti-TNF option in IBD. The gastroenterology community’s familiarity with golimumab in UC and the convenience of monthly subcutaneous dosing keeps it relevant even amid broader market pressure.
What Patients Should Know Right Now
If you are currently on Simponi and well-controlled, your treatment is unaffected by the LOE dynamics. The drug is available, Janssen is still manufacturing it, and your prescriber should be managing your care as usual. The absence of a commercially available biosimilar is, paradoxically, the most stable situation for a currently treated patient: there is no formulary switch coming in the near term.
If you are facing cost barriers with Simponi: Janssen’s patient assistance program and specialty pharmacy support are the primary access pathways available now. The HealthWell Foundation, Patient Advocate Foundation, and The Assistance Fund also provide copay assistance for biologics in autoimmune disease.
If you are being newly evaluated for a TNF inhibitor: the choice between golimumab and the available adalimumab biosimilars, which are often preferred by payers, should be made with your rheumatologist or gastroenterologist based on your specific disease, prior treatment history, and how your insurance formulary is structured.
The biosimilar landscape for Simponi will clarify over the next 12 to 24 months as Alvotech addresses the manufacturing deficiencies cited in its CRL and as the Immgolis litigation works through the courts. When an approved, commercially available golimumab biosimilar does launch in the U.S., it will enter a market that already has strong biosimilar momentum from the adalimumab experience, meaning the conversion dynamics and price competition could move faster than they did for earlier biologic LOEs.
For related HED coverage on how the BPCIA patent litigation process works and what it means when an approved biosimilar is blocked from launch, see our dedicated post on Immgolis and Immgolis Intri and our post on PONLIMSI and why biosimilar FDA approval does not automatically translate to patient savings.
Sources
Simponi FDA approval: FDA approves golimumab (Simponi). FDA.gov.
Simponi Aria FDA approval: FDA approves golimumab (Simponi Aria). FDA.gov.
Optum LOE overview: Blockbuster drug patent expirations in 2026 and what they mean. business.optum.com. April 2026.
AVT05 CRL (Alvotech): Alvotech Provides Update on the Status of U.S. BLA for AVT05. GlobeNewswire. November 2, 2025.
AVT05 CRL analysis: FDA Issues CRL for Alvotech’s Simponi Biosimilar AVT05. PearceIP. November 2025.
BAT2506/Immgolis FDA approval and BPCIA litigation: FDA approves first interchangeable biosimilars to Simponi and Simponi Aria. FDA.gov. May 15, 2026. | Janssen Files First BPCIA Suit Over Simponi Biosimilar. BiologicsHQ. March 2026.
HED Immgolis post: The First Biosimilars to Simponi and Simponi Aria Just Got FDA Approval. healthevidencedigest.com.
Golimumab mechanism and indications (StatPearls): Golimumab. StatPearls. NCBI.
TNF inhibitor safety overview (StatPearls): Tumor Necrosis Factor Inhibitors. StatPearls. NCBI.
TNF-alpha biology: Tumor Necrosis Factor. StatPearls. NCBI.
GO-FORWARD trial primary publication: Keystone EC et al. Golimumab in patients with active RA despite methotrexate therapy (GO-FORWARD). Ann Rheum Dis. 2009;68(6):789–796.
GO-RAISE trial (ankylosing spondylitis): Inman RD et al. Efficacy and safety of golimumab in patients with ankylosing spondylitis (GO-RAISE). Arthritis Rheum. 2008;58(11):3402–3412.
GO-REVEAL trial (psoriatic arthritis): Kavanaugh A et al. Golimumab in patients with active PsA (GO-REVEAL). Ann Rheum Dis. 2009;68(4):498–505.
Five-year persistence data: Weinstein CLJ et al. Long-term golimumab persistence: five-year treatment retention data. Clin Rheumatol. 2023;42(12):3397.
PURSUIT-SC and Maintenance trials: Sandborn WJ et al. Subcutaneous golimumab induces clinical response and remission in moderate-to-severe ulcerative colitis (PURSUIT). Gastroenterology. 2014;146:85–95.
TB risk with golimumab: Cantini F et al. Tuberculosis risk with recently licensed TNF-alpha inhibitors. J Rheumatol Suppl. 2014. PMID 24789001.
Latent TB testing before biologics: Testing for Latent TB Infection. CDC.
Simponi prescribing information: Simponi (golimumab) Prescribing Information. Janssen Biotech.
NIAMS disease overviews: Rheumatoid Arthritis | Psoriatic Arthritis | Ankylosing Spondylitis
NIDDK ulcerative colitis: Ulcerative Colitis. niddk.nih.gov.
Patient resources: Arthritis Foundation | Crohn’s and Colitis Foundation | Simponi patient support | HealthWell Foundation | Patient Advocate Foundation | The Assistance Fund
| Disclaimer: Health Evidence Digest provides general information about FDA approvals, loss of exclusivity events, and health research for educational purposes. This content is not a substitute for professional medical advice. Decisions about TNF inhibitor therapy, including golimumab, require individualized assessment by a board-certified rheumatologist, gastroenterologist, or other appropriate specialist, accounting for the patient’s complete medical history, infection risk profile, vaccination status, and concurrent medications. Never discontinue a biologic therapy without medical guidance. |
|---|