| 📌 The essentials On May 29, 2026, the FDA approved Zaynich (cefepime and zidebactam, Wockhardt) for the treatment of adults with complicated urinary tract infections (cUTI), including pyelonephritis, caused by susceptible Gram-negative pathogens. What makes it mechanistically distinctive: zidebactam is a non-beta-lactam beta-lactamase inhibitor that acts as a beta-lactam enhancer, functioning both as a beta-lactamase inhibitor and as an independent antibacterial agent targeting PBP2. This dual role allows the combination to overcome multiple resistance mechanisms including carbapenem-hydrolyzing metallo-beta-lactamases (MBLs), which no currently approved beta-lactam/beta-lactamase inhibitor combination can address. Approved organisms: Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, Enterobacter cloacae complex, and Pseudomonas aeruginosa. The clinical basis: Phase 3 ENHANCE-1 trial (NCT04979806), 530 patients, double-blind, randomized 2:1 Zaynich versus meropenem. Primary endpoint: composite clinical cure and microbiologic response rate 89.0% with Zaynich versus 68.4% with meropenem (difference +20.6%; 95% CI 12.3 to 29.5). Non-inferiority and superiority both achieved. Dosing: 3 g (cefepime 2 g plus zidebactam 1 g) intravenous infusion every 8 hours for 7 to 10 days, infused over 1 hour. Dose adjustment required for renal impairment. Regulatory designations: Qualified Infectious Disease Product (QIDP), Fast Track. Global status: also approved in India May 27, 2026; EMA Marketing Authorization Application submitted. |
|---|
Complicated urinary tract infections are not the same as the straightforward UTIs that can be treated with a few days of oral antibiotics. They involve the upper urinary tract (pyelonephritis, kidney infection), occur in patients with structural or functional urinary tract abnormalities, affect hospitalized patients or those with indwelling urinary catheters, or are caused by pathogens that resist the antibiotics typically used for uncomplicated infections. In the United States, cUTIs account for more than 600,000 hospitalizations annually, and the proportion caused by multidrug-resistant (MDR) Gram-negative bacteria is growing.
The antimicrobial resistance crisis in Gram-negative bacteria is not a future problem. It is a present clinical reality. Carbapenem-resistant Enterobacteriaceae (CRE) and carbapenem-resistant Pseudomonas aeruginosa are among the pathogens designated by the CDC as urgent threats. Carbapenems, the last-resort antibiotics for many Gram-negative infections, are failing against an expanding proportion of resistant organisms. New antibiotics that can overcome carbapenem resistance mechanisms, particularly metallo-beta-lactamases, are a clinical priority.
On May 29, 2026, the FDA approved Zaynich (cefepime and zidebactam), the first approved antibiotic combination in which the beta-lactamase inhibitor component also functions as an independent antibacterial agent. The ENHANCE-1 trial showed not only non-inferiority to meropenem (a carbapenem and the current gold standard for complicated Gram-negative infections) but superiority: a 20.6 percentage point higher composite cure rate. For an antibiotic trial, that is a striking result.
The Antimicrobial Resistance Context: Why New Gram-Negative Antibiotics Matter
Antimicrobial resistance in Gram-negative bacteria operates through several distinct mechanisms that have made treatment of serious infections increasingly challenging.
Beta-lactamase enzymes: the central resistance mechanism
Beta-lactamases are enzymes produced by bacteria that break down the beta-lactam ring, the core structure of penicillins, cephalosporins, and carbapenems. By hydrolyzing the beta-lactam ring, these enzymes inactivate the antibiotic before it can reach its target (penicillin-binding proteins, or PBPs) on the bacterial cell wall.
The major beta-lactamase classes relevant to Gram-negative resistance include:
- Extended-spectrum beta-lactamases (ESBLs): Inactivate most penicillins and cephalosporins. Very common in E. coli and Klebsiella. Previously manageable with carbapenems.
- KPC-type carbapenemases (Klebsiella pneumoniae carbapenemase): Inactivate carbapenems. Now widespread in Klebsiella, Enterobacter, and other Enterobacterales.
- Metallo-beta-lactamases (MBLs) including NDM, VIM, and IMP: Also inactivate carbapenems. Critically, unlike serine-based carbapenemases, MBLs use a zinc-dependent mechanism that conventional serine-beta-lactamase inhibitors (clavulanate, tazobactam, avibactam) cannot block.
The MBL gap is the most clinically urgent. Approved combinations such as ceftazidime-avibactam (Avycaz) and meropenem-vaborbactam (Vabomere) are effective against KPC-producing organisms but cannot inhibit MBL-producing bacteria. For patients with MBL-producing infections, treatment options have been extremely limited.
Why Pseudomonas aeruginosa is a separate challenge
Pseudomonas aeruginosa is an intrinsically resistant Gram-negative bacterium that combines multiple resistance mechanisms: it produces beta-lactamases, it can upregulate efflux pumps that expel antibiotics from the cell, and it can downregulate outer membrane porins that antibiotics use to enter. Many standard cephalosporins have limited activity against Pseudomonas; cefepime has historically been among the more active cephalosporins against this pathogen, making it a rational backbone for the Zaynich combination.
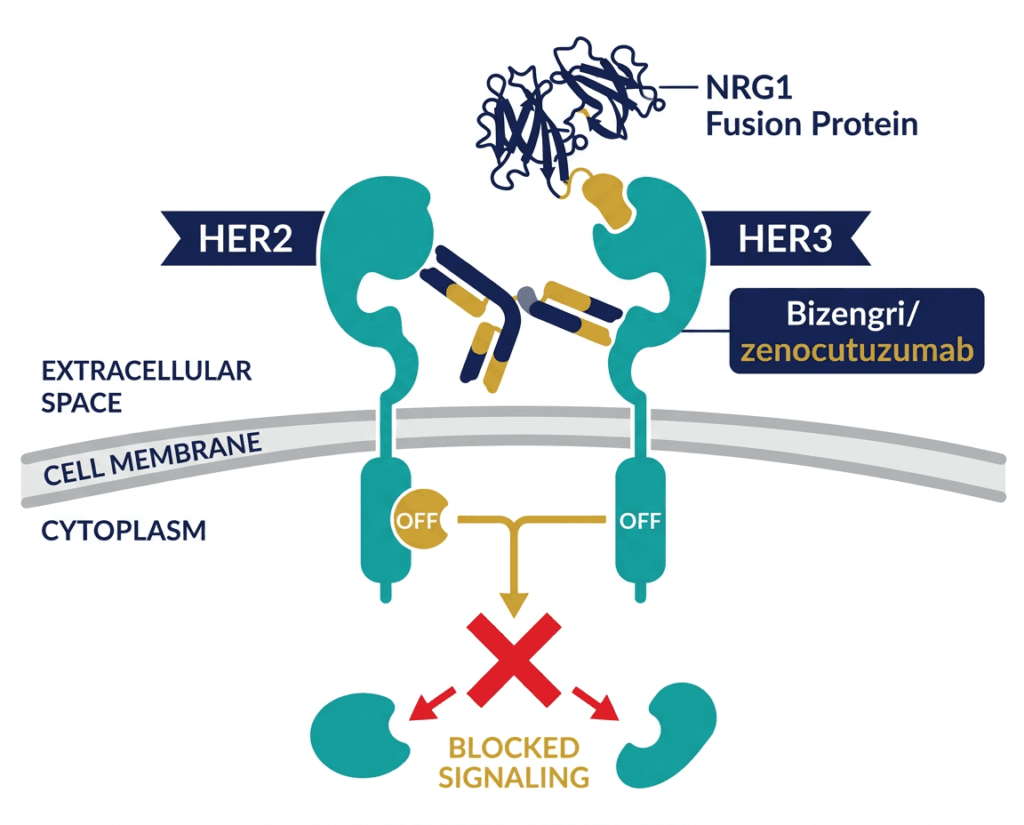
| Why zidebactam is different from all previously approved beta-lactamase inhibitors All previously approved beta-lactamase inhibitors (clavulanate, sulbactam, tazobactam, avibactam, vaborbactam, relebactam) work by inhibiting beta-lactamase enzymes, preventing them from destroying the antibiotic partner. Zidebactam does this too, but it also independently targets PBP2 in Gram-negative bacteria, the same penicillin-binding protein targeted by carbapenems. By directly occupying PBP2, zidebactam restores susceptibility to cefepime in bacteria that have become resistant through beta-lactamase production, because even if some residual beta-lactamase activity breaks down cefepime molecules, zidebactam’s independent PBP2 targeting contributes directly to bacterial cell wall disruption. This beta-lactam enhancer mechanism is what makes Zaynich active against MBL-producing organisms where avibactam-containing combinations fail. Avibactam cannot inhibit MBLs; zidebactam bypasses the need to inhibit them. |
|---|
How Zaynich Works: The Dual Mechanism in Detail
Zaynich combines two drugs that act synergistically through distinct but complementary mechanisms targeting the Gram-negative bacterial cell wall:
Cefepime: the fourth-generation cephalosporin
Cefepime is a fourth-generation cephalosporin with good activity against both Gram-negative and some Gram-positive organisms. In Enterobacterales, cefepime primarily targets PBP3 and PBP1a/1b. In Pseudomonas aeruginosa, it targets PBP3. PBP binding inhibits cell wall synthesis, ultimately causing bacterial lysis. Cefepime’s advantage over earlier cephalosporins is its stability against many (but not all) beta-lactamases and its enhanced ability to penetrate Gram-negative outer membranes.
Zidebactam: the beta-lactam enhancer
Zidebactam is a novel synthetic molecule classified as a diazabicyclooctane (DBO), structurally similar to avibactam and relebactam. Its two mechanisms work simultaneously:
As a beta-lactamase inhibitor: Zidebactam inhibits serine-beta-lactamases including Class A (ESBLs, KPC), Class C (AmpC), and Class D (OXA) beta-lactamases, protecting cefepime from enzymatic degradation. Importantly, it is also active against several serine-carbapenemases, extending coverage to KPC-producing organisms.
As a direct PBP2 targeting agent: Zidebactam binds PBP2 with high affinity in Gram-negative pathogens. This independent bactericidal action contributes to cell wall disruption independent of its beta-lactamase inhibitory role. For MBL-producing organisms (NDM, VIM, IMP), where the MBL enzyme cannot be inhibited by any current serine-targeting inhibitor, zidebactam’s PBP2 binding provides direct antibacterial activity that partially compensates for ongoing cefepime destruction by the MBL. This is the mechanism that gives the combination activity against MBL producers.
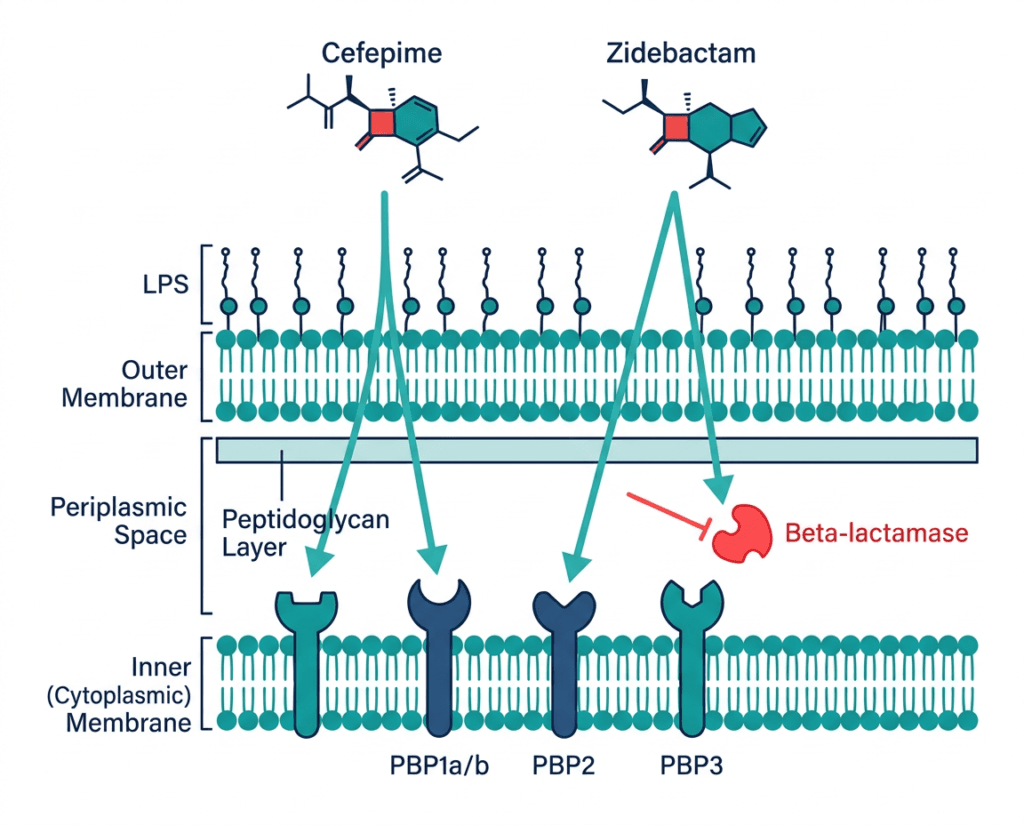
The combined effect is coverage of PBP1a/1b, PBP2, and PBP3 simultaneously in the majority of clinically relevant Gram-negative pathogens, across diverse resistance mechanisms.
The ENHANCE-1 Trial: Full Results
Design
ENHANCE-1 (NCT04979806) was a Phase 3, randomized, double-blind, multicenter, non-inferiority trial. The trial enrolled 530 hospitalized adults at 64 clinical sites in the United States, Europe, Latin America, China, and India.
Eligible patients: Adults aged 18 and older with a clinical diagnosis of cUTI or acute pyelonephritis, with or without concurrent bacteremia, requiring intravenous antibiotic therapy.
Randomization: 2:1 to:
- Zaynich: cefepime 2 g plus zidebactam 1 g (total 3 g) by 1-hour IV infusion every 8 hours for 7 to 10 days (n=352)
- Meropenem: 1 g by 30-minute IV infusion every 8 hours for 7 to 10 days (n=177)
Dose adjustments for renal function were specified per protocol for both arms.
Primary endpoint: Composite of clinical cure and microbiologic response at the test-of-cure (TOC) visit (approximately 7 to 14 days after end of treatment) in the modified microbiological intent-to-treat (mMITT) population.
Non-inferiority margin: The trial was powered for non-inferiority but also had a pre-specified superiority analysis.
Primary endpoint results
| Outcome at test-of-cure visit | Zaynich (n=352) | Meropenem (n=177) | Treatment difference |
|---|---|---|---|
| Composite clinical cure and microbiologic response rate | 89.0% | 68.4% | +20.6% (95% CI 12.3 to 29.5) |
| Non-inferiority achieved | Yes | — | Confidence interval lower bound far above zero |
| Superiority achieved | Yes | — | Lower bound of 95% CI (12.3%) above zero |
Source: ENHANCE-1, NCT04979806. Presented at IDSA 2025 Open Forum. Wockhardt press release. June 1, 2026.
The 20.6 percentage point superiority over meropenem in composite clinical and microbiologic response deserves specific comment. Meropenem is not a weak comparator. It is among the broadest-spectrum, most reliably active intravenous antibiotics available for Gram-negative cUTI, and it was the appropriate comparator given the spectrum of Gram-negative organisms targeted. An antibiotic demonstrating superiority over meropenem in a well-conducted Phase 3 trial is an unusual and clinically meaningful result.
The likely explanation lies in the patient population and organism mix: a proportion of patients in the trial carried organisms with resistance mechanisms that meropenem could not fully overcome but that Zaynich’s broader mechanism could address. The trial’s global enrollment across geographies with high burdens of ESBL-producing and carbapenem-resistant organisms supports this interpretation.
Safety
Zaynich was generally well tolerated in ENHANCE-1. The most common adverse reactions, occurring in at least 2% of Zaynich-treated patients, were diarrhea, hypertension, headache, and hypokalemia. These are consistent with the known safety profiles of the individual components.
Cefepime-specific safety considerations:
- Neurotoxicity: cefepime is associated with neurotoxic effects including encephalopathy, myoclonus, and seizures, particularly at high doses or in patients with renal impairment receiving unadjusted doses. The prescribing information specifically warns that serious neurologic adverse reactions have occurred in geriatric patients with renal insufficiency given unadjusted doses of cefepime.
- Hypersensitivity reactions: including anaphylaxis, consistent with the cephalosporin class.
- Clostridioides difficile-associated diarrhea (CDAD): as with all antibiotics, C. diff colitis is a risk with broad-spectrum agents.
Approved Organisms and the Susceptibility Testing Requirement
Zaynich is approved specifically for cUTI caused by the following susceptible organisms, as determined by appropriate microbiologic testing:
- Escherichia coli
- Klebsiella pneumoniae
- Proteus mirabilis
- Enterobacter cloacae complex
- Pseudomonas aeruginosa
The word “susceptible” is clinically operative. Zaynich should be used when the infecting organism has been confirmed or is strongly suspected to be one of these organisms based on culture and susceptibility data. Empirical use without culture data should follow local epidemiology and institutional antibiotic stewardship guidance.
The approved coverage does not include Gram-positive organisms, anaerobes, or Acinetobacter species. For patients with polymicrobial infections or suspected Gram-positive co-infection, additional agents may be required.
Dosing and Administration
| Parameter | Details |
|---|---|
| Standard dose | 3 g (cefepime 2 g plus zidebactam 1 g) IV every 8 hours |
| Infusion duration | 1 hour per infusion |
| Treatment duration | 7 to 10 days |
| Renal dose adjustment | Required for eGFR below 50 mL/min per 1.73m²; see full prescribing information |
| Geriatric patients | Extra caution; monitor renal function and adjust dose accordingly given cefepime neurotoxicity risk |
| Preparation | Sterile powder reconstituted per prescribing information instructions before IV infusion |
Regulatory Designations and Context
Zaynich received two FDA designations reflecting the urgency of the antimicrobial resistance problem it addresses:
Qualified Infectious Disease Product (QIDP): A designation created by the GAIN Act (Generating Antibiotic Incentives Now) specifically to incentivize development of antibiotics against serious or life-threatening infections, including drug-resistant Gram-negative bacteria. QIDP status provides 5 additional years of market exclusivity, priority review, and fast track eligibility.
Fast Track designation: Provides more frequent FDA interactions during development and eligibility for rolling review.
Globally, Zaynich was also approved by the Drugs Controller General of India (DCGI) on May 27, 2026, two days before U.S. approval. Wockhardt has submitted a Marketing Authorization Application (MAA) to the European Medicines Agency (EMA). The drug was made available through expanded access programs in multiple countries before formal approval.
Where Zaynich Fits in the Antibiotic Landscape for Resistant Gram-Negative cUTI
The antibiotic options for MDR Gram-negative cUTI have expanded modestly in recent years, with each agent covering a different subset of resistance mechanisms:
| Agent | Covers ESBLs | Covers KPC | Covers MBLs (NDM/VIM/IMP) | Covers P. aeruginosa |
|---|---|---|---|---|
| Meropenem (carbapenem) | Yes | No (KPC destroys it) | No | Yes |
| Ceftazidime-avibactam (Avycaz) | Yes | Yes | No | Yes |
| Meropenem-vaborbactam (Vabomere) | Yes | Yes | No | Limited |
| Imipenem-cilastatin-relebactam (Recarbrio) | Yes | Yes | No | Yes |
| Ceftolozane-tazobactam (Zerbaxa) | Yes (limited) | No | No | Enhanced P. aeruginosa coverage |
| Zaynich (cefepime-zidebactam) | Yes | Yes | Partial (via PBP2 mechanism) | Yes |
The MBL coverage column is the most important for clinical decision-making. For patients with infections confirmed or strongly suspected to be caused by NDM-producing, VIM-producing, or IMP-producing organisms, Zaynich’s PBP2-targeting mechanism provides an option where no previously approved combination is reliably effective. This is the niche where Zaynich represents the most meaningful advance.
For general ESBL-producing and KPC-producing infections, the clinical choice between Zaynich, ceftazidime-avibactam, meropenem-vaborbactam, or imipenem-relebactam will depend on local susceptibility patterns, institutional formulary decisions, and patient-specific factors.
Antibiotic Stewardship Considerations
Zaynich is a broad-spectrum intravenous antibiotic approved for a specific clinical indication. Its role should be guided by antimicrobial stewardship principles:
- Use based on culture and susceptibility data whenever possible
- Employ empirically when MDR pathogens are strongly suspected based on prior culture history, recent antibiotic exposure, healthcare-associated acquisition, or local epidemiology
- De-escalate to narrower-spectrum agents when susceptibility data permit
- Follow institutional stewardship guidelines and consult infectious disease specialists for complex MDR cases
For related HED coverage on the regulatory framework that incentivizes antibiotic development and on drug safety monitoring for serious infections, see our post on Hepcludex (bulevirtide) as the first approved treatment for hepatitis delta and our post on Tavneos (avacopan) and the serious liver injury warning in post-market surveillance.
Sources
FDA approval / Wockhardt press release: Wockhardt Receives U.S. FDA Approval for ZAYNICH (cefepime and zidebactam). PRNewswire. June 1, 2026.
Drugs.com approval news: FDA Approves Zaynich (cefepime and zidebactam) for the Treatment of Complicated Urinary Tract Infections. drugs.com. May 30, 2026.
Urology Times clinical coverage: FDA approves cefepime and zidebactam for complicated UTI. urologytimes.com. May 2026.
Renal and Urology News: Zaynich Approved for Complicated Urinary Tract Infections in Adults. renalandurologynews.com. June 2026.
Contemporary OB/GYN: FDA approves cefepime and zidebactam intravenous antibiotic to treat adults with cUTI. contemporaryobgyn.net. June 2026.
ENHANCE-1 IDSA 2025 Open Forum presentation: Genov P, Mladenov B, Slaitas D, et al. Efficacy of beta-lactam enhancer based zidebactam-cefepime combination (WCK 5222) versus meropenem in adults with cUTI or acute pyelonephritis: global, randomized, double-blind, Phase 3 trial. Open Forum Infectious Diseases. 2026;ofaf695.1402. doi:10.1093/ofid/ofaf695.1402.
ENHANCE-1 trial registration: NCT04979806. ClinicalTrials.gov.
Zaynich full prescribing information: ZAYNICH (cefepime and zidebactam) Prescribing Information. Wockhardt. 2026.
Beta-lactamases and resistance mechanisms: Beta-Lactamase Inhibitors. PMC7279573.
Complicated UTI overview: Complicated Urinary Tract Infections. StatPearls. NCBI.
AMR in Gram-negative bacteria: Gram-Negative Bacterial Infections. StatPearls. NCBI.
Pseudomonas aeruginosa: Pseudomonas aeruginosa Infections. StatPearls. NCBI.
Cefepime neurotoxicity: Cefepime-Induced Neurotoxicity. PMC7459434.
Cefepime mechanism: Cefepime. StatPearls. NCBI.
Cephalosporin hypersensitivity: Cephalosporin Allergy. StatPearls. NCBI.
C. diff associated diarrhea: Clostridioides Difficile. StatPearls. NCBI.
CDC AMR urgent threats: CDC Antibiotic Resistance Threats Report. cdc.gov.
CDC antibiotic stewardship core elements: Core Elements of Antibiotic Stewardship. cdc.gov.
QIDP and GAIN Act: GAIN Act Frequently Asked Questions. FDA.gov.
Fast Track designation: Fast Track. FDA.gov.
| Disclaimer: Health Evidence Digest provides general information about FDA approvals and health research for educational purposes. This content is not a substitute for professional medical advice, diagnosis, or treatment. Zaynich is an intravenous antibiotic requiring prescription and clinical oversight. Decisions about antibiotic selection for complicated urinary tract infections should be made by a qualified clinician based on culture and susceptibility data, patient-specific factors including renal function, and institutional antimicrobial stewardship guidelines. |
|---|