| 📌 The essentials On June 12, 2026 (announced June 13), the FDA granted accelerated approval to Tzield (teplizumab-mzwv, Sanofi) for a new indication: to delay the decline in endogenous (the patient’s own) insulin production in pediatric patients aged 8 through 17 years recently diagnosed with Stage 3 type 1 diabetes (T1D). This is the first FDA-approved disease-modifying therapy for recently diagnosed Stage 3 type 1 diabetes. Prior to this approval, there was no treatment capable of altering the course of T1D after clinical diagnosis. Critical time window: treatment must be initiated within 8 weeks of Stage 3 T1D diagnosis. This is an acute clinical opportunity: families and pediatric endocrinologists need to be aware that this window exists and act accordingly. Regulatory pathway: accelerated approval based on C-peptide (a surrogate marker of beta-cell function reasonably likely to predict clinical benefit). A post-approval confirmatory study, the Phase 3 BETA-PRESERVE trial, is ongoing. The clinical basis: Phase 3 PROTECT trial (NCT03875729), 328 youth with Stage 3 T1D enrolled within 6 weeks of diagnosis. Two 12-day IV infusion courses (at baseline and at week 26). Primary endpoint: stimulated C-peptide AUC at 78 weeks, a validated surrogate of beta-cell function. Teplizumab significantly preserved C-peptide versus placebo at 78 weeks. Clinically meaningful beta-cell function maintained in 94.9% of teplizumab-treated patients versus 79.2% of controls. Supporting outcomes: better HbA1c, more time-in-range, less insulin use, and lower hypoglycemia favored teplizumab numerically, though without statistical significance for each. Tzield is not effective as a disease-modifying therapy in non-autoimmune dysglycemic conditions: it only works when the underlying cause is immune-mediated beta-cell destruction. Key safety warnings: cytokine release syndrome (CRS), lymphopenia, viral reactivation including EBV and CMV (most serious cases in patients who continued treatment despite persistent severe lymphopenia). Tzield’s complete indication picture after June 2026: Stage 2 T1D (delay of Stage 3 onset) in patients aged 1 year and older; Stage 3 T1D (delay of insulin production decline) in patients aged 8 to 17 years recently diagnosed. |
|---|
Type 1 diabetes is an autoimmune disease in which the immune system destroys the insulin-producing beta cells of the pancreas. For almost the entirety of its known history, nothing in medicine could change that course after it began. Insulin was discovered in 1921 and saved countless lives, but it did not address the underlying immune attack. Immunosuppressive agents reduced beta-cell destruction in small studies but caused unacceptable side effects. The disease progressed. Eventually, the beta cells were gone.
Teplizumab (Tzield) was the first drug to change any part of that story. Its original approval in November 2022 for Stage 2 T1D (the pre-symptomatic stage, before diagnosis) demonstrated that a 14-day course of IV treatment could delay the onset of clinical diabetes by approximately two years in at-risk individuals. That was extraordinary. But it left open a question: what about the children who were already diagnosed?
On June 12, 2026, the FDA answered that question. Tzield received accelerated approval for use after diagnosis, specifically to delay the decline of the patient’s own insulin production in children and adolescents aged 8 to 17 who have been recently diagnosed with Stage 3 T1D. It is the first disease-modifying therapy ever approved for recently diagnosed type 1 diabetes. There has never been anything like it before.
This post covers the staged model of type 1 diabetes and why it matters clinically, how teplizumab’s CD3 mechanism works to modulate the autoimmune attack, what the PROTECT trial showed, what the accelerated approval pathway means for the evidence base, what the 8-week treatment window requires from families and clinicians, and what this approval means for the type 1 diabetes community.
The Staged Model of Type 1 Diabetes: Why the Stage Matters
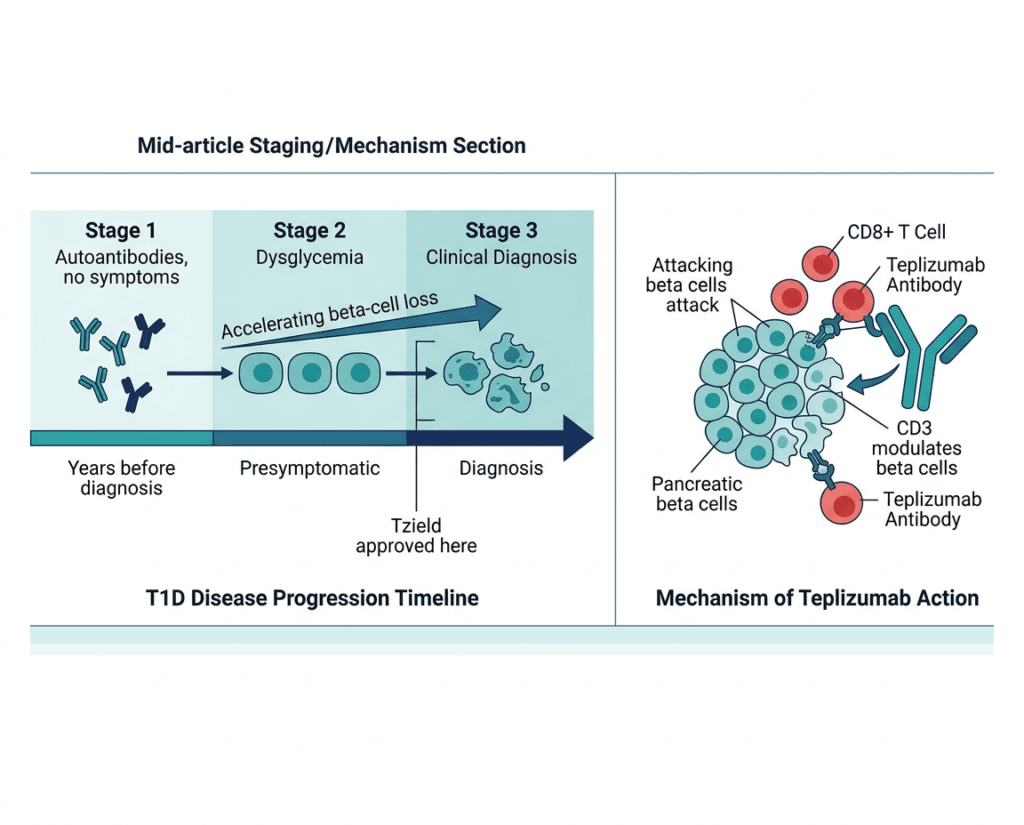
Type 1 diabetes is not a single event. It is a continuum of progressive autoimmune destruction of pancreatic beta cells that begins years before clinical diagnosis and continues afterward. The staged model, developed by the American Diabetes Association and JDRF (now Breakthrough T1D), provides a framework that has reshaped how clinicians and researchers understand and now treat the disease.
| Stage | Definition | What it means clinically |
|---|---|---|
| Stage 1 T1D | Two or more positive islet autoantibodies; normoglycemia; no symptoms | Beta-cell autoimmunity established; at high risk for progression; no metabolic dysfunction yet |
| Stage 2 T1D | Two or more positive islet autoantibodies; dysglycemia (glucose abnormalities without meeting diabetes criteria); no symptoms | Beta-cell loss accelerating; progression to Stage 3 virtually certain without intervention; Tzield approved here (Stage 2 in patients aged 1 year and older) |
| Stage 3 T1D | Two or more positive autoantibodies plus symptomatic hyperglycemia meeting diabetes diagnostic criteria | Clinical diabetes: the stage families recognize as “diagnosis.” Beta-cell function still partially preserved at this point; Tzield now approved here in patients aged 8 to 17 years recently diagnosed |
The critical insight embedded in this staging model, and the reason the June 2026 approval matters so much, is that Stage 3 T1D does not begin with zero remaining beta cells. At the time of clinical diagnosis, a meaningful fraction of insulin-producing capacity is still present. The autoimmune attack has been ongoing for years, but it has not yet completed its work. There is a window, measured in weeks to months after clinical diagnosis, during which intervention to slow or interrupt the immune destruction of the remaining beta cells is still biologically possible and clinically meaningful.
That window is exactly what teplizumab’s new indication targets.
| Why preserving residual beta-cell function matters clinically Even partial preservation of the patient’s own insulin production, measured as C-peptide secretion, translates into tangible clinical advantages that insulin therapy alone cannot replicate. Patients with measurable residual beta-cell function consistently show better HbA1c levels, more time in glycemic range, lower insulin requirements, and meaningfully reduced frequency of severe hypoglycemia compared to patients with no endogenous insulin production. The pancreas’s own insulin secretion is exquisitely responsive to real-time blood glucose fluctuations in a way that no external insulin replacement strategy, including advanced closed-loop systems, can fully replicate. This is the clinical rationale for C-peptide preservation as both a trial endpoint and a treatment goal: it is not an abstract biomarker. It is a direct predictor of quality of life, safety, and long-term diabetes outcomes. |
|---|
How Teplizumab Works: The CD3 Mechanism
Teplizumab is a humanized monoclonal antibody that targets CD3, a protein complex on the surface of T cells. Understanding why blocking CD3 helps preserve beta cells requires a brief look at the immunology of type 1 diabetes.
In type 1 diabetes, autoreactive CD8+ cytotoxic T cells and CD4+ helper T cells infiltrate the pancreatic islets and selectively destroy the beta cells that produce insulin. These T cells recognize beta-cell antigens (such as glutamic acid decarboxylase and insulin itself) as foreign and mount a sustained immune attack. The attack is not a single event; it is an ongoing, progressive destruction that continues over years and accelerates toward the time of clinical diagnosis and beyond.
CD3 is a component of the T-cell receptor complex: it is the intracellular signaling module that transduces the activation signal when a T cell’s antigen receptor binds its target. By binding CD3, teplizumab modulates T-cell activation without completely eliminating T cells from the circulation. This partial modulation is important: unlike conventional immunosuppressants that broadly suppress all T cell activity (creating broad susceptibility to infection), teplizumab’s mechanism selectively tolerizes the autoreactive T cells most responsible for beta-cell destruction while preserving broader immune function.
The proposed mechanism includes several complementary effects: induction of partially exhausted or tolerogenic T cells that suppress the autoimmune response; preferential depletion of effector T cells in the inflamed islets; and expansion of regulatory T cells that actively suppress autoimmunity. The net result, observed in both the original Stage 2 trials and now in the Stage 3 PROTECT trial, is a slower rate of beta-cell destruction measurable as preserved C-peptide secretion over years of follow-up after the treatment is complete.
Teplizumab is administered as a 14-day intravenous infusion course, not as an ongoing therapy. Two courses are given: one at baseline (treatment initiation) and one at week 26 (6 months later). After that, no further doses are required. The immune reprogramming established by the two courses appears to produce durable benefit, as evidenced by the persistent C-peptide preservation observed in PROTECT through 78 weeks after enrollment.
The PROTECT Trial: What the Evidence Shows
Design
PROTECT (NCT03875729) was a Phase 3, randomized, double-blind, placebo-controlled multinational trial specifically designed to evaluate teplizumab in newly diagnosed Stage 3 T1D. The trial enrolled 328 children and adolescents (teplizumab n=217, placebo n=111) within 6 weeks of Stage 3 T1D diagnosis.
Treatment regimen: Two 12-day intravenous infusion courses of teplizumab or placebo: one at baseline and one at week 26, in addition to standard diabetes care (insulin therapy and glucose monitoring).
Primary endpoint: Stimulated C-peptide area under the curve (AUC) at 78 weeks, measured during a mixed meal tolerance test (MMTT). C-peptide is co-secreted with insulin from beta cells; its level in the blood is a direct measure of functional beta-cell mass, unaffected by exogenous insulin administration.
Primary endpoint results
Teplizumab significantly preserved stimulated C-peptide versus placebo at 78 weeks. C-peptide is a validated surrogate endpoint for beta-cell function that is reasonably likely to predict clinical benefit, which is the evidentiary standard for accelerated approval.
Secondary and supporting outcomes
| Outcome | Teplizumab | Placebo | Notes |
|---|---|---|---|
| Clinically meaningful beta-cell function maintained at 78 weeks | 94.9% | 79.2% | Clinically significant difference; higher proportion preserved residual function |
| HbA1c | Numerically favored teplizumab | Reference | Did not reach statistical significance |
| Time in range | Numerically favored teplizumab | Reference | Did not reach statistical significance |
| Insulin dose | Numerically lower with teplizumab | Reference | Did not reach statistical significance |
| Hypoglycemia | Numerically lower with teplizumab | Reference | Did not reach statistical significance |
Source: Ramos EL, Dayan CM, Chatenoud L, et al. Teplizumab and beta-cell function in newly diagnosed type 1 diabetes. NEJM. 2023;389(23):2151–2161. doi:10.1056/NEJMoa2306691. PROTECT NCT03875729.
The finding that 94.9% of teplizumab-treated patients maintained clinically meaningful beta-cell function compared to 79.2% of controls at 78 weeks is clinically important. It means that a substantially higher proportion of treated children preserved enough of their own insulin production to realize the downstream glycemic benefits associated with residual beta-cell function.
The secondary outcome numerical trends (better HbA1c, more time in range, less insulin needed, less hypoglycemia) are directionally consistent with what prior research has shown about the clinical value of C-peptide preservation, even without reaching individual statistical significance. They support the clinical plausibility of the C-peptide surrogate endpoint.
Dr. Kevan Herold, MD, C.N.H. Long Professor of Immunobiology and Medicine at Yale School of Medicine and co-author of the PROTECT trial, has stated that practically all patients with new-onset Stage 3 T1D should be offered therapy unless there is an underlying condition that would prevent drug administration, describing the absence of other disease-modifying alternatives for T1D as the defining context for this recommendation.
The accelerated approval basis and confirmatory study
The FDA granted this approval under the accelerated approval pathway, based on C-peptide as a surrogate endpoint reasonably likely to predict clinical benefit. This is the same pathway used for many rare disease and serious condition approvals where waiting for long-term clinical outcome data would delay access to potentially beneficial therapies.
The BETA-PRESERVE trial (NCT05902884), a Phase 3 confirmatory study, is ongoing to verify the clinical benefit of C-peptide preservation in terms of direct patient outcomes including HbA1c, time in range, insulin requirements, and hypoglycemia frequency. Continued approval is contingent on the confirmatory study verifying this clinical benefit.
The 8-Week Treatment Window: What This Means Practically
The approved indication specifies patients recently diagnosed with Stage 3 T1D. Breakthrough T1D has clarified this as within the last 8 weeks of Stage 3 diagnosis. This is not an administrative convenience: it is a biological reality. The earlier treatment is initiated after clinical diagnosis, the more residual beta-cell function remains to preserve. Waiting months after diagnosis substantially reduces the potential benefit.
This 8-week window creates a specific and urgent clinical requirement: families and pediatric endocrinologists must be aware of Tzield’s availability at the time of diagnosis, not months later. For families receiving a new T1D diagnosis in a child or adolescent, this means:
- Ask the diagnosing pediatric endocrinologist at the initial appointment whether Tzield is appropriate for your child
- If your child is aged 8 to 17, was diagnosed within the past 8 weeks, and has confirmed autoimmune T1D, the conversation about Tzield should happen now, not at the 3-month follow-up visit
- A referral to an academic center or JDRF-connected care team with experience in disease-modifying therapy may be appropriate if the diagnosing center is not yet familiar with the treatment protocol
The two 12-day IV infusion courses require IV access and monitoring during infusions, which means this is not a home therapy. It requires an infusion center or hospital setting, coordination of insurance authorization, and scheduling. These logistics take time, making awareness at diagnosis critical for staying within the treatment window.
Tzield’s Complete Indication Picture After June 2026
Two approvals and one significant expansion have occurred in 2026:
| Indication | Population | Approval date |
|---|---|---|
| Delay onset of Stage 3 T1D | Adults and pediatric patients aged 1 year and older with Stage 2 T1D | November 2022 (original); expanded to ages 1 to 7 in April 2026 (previously 8 and older) |
| Delay decline in insulin production in recently diagnosed Stage 3 T1D | Pediatric patients aged 8 to 17 years within 8 weeks of Stage 3 T1D diagnosis | June 12, 2026 (accelerated approval) |
The Stage 2 expansion to children as young as 1 year (April 2026, based on PETITE-T1D study data) and the Stage 3 approval (June 2026, PROTECT) together mean that teplizumab is now available across the full spectrum of high-risk and newly diagnosed pediatric T1D, from presymptomatic at-risk toddlers through adolescents at clinical diagnosis.
Safety: What the Prescribing Information and Trial Data Show
Teplizumab’s safety profile is driven by its mechanism of CD3-directed T-cell modulation during 12-day IV infusion courses. The adverse event profile in PROTECT was consistent with the previously characterized teplizumab dataset from over 900 patients across the development program.
Warnings and key adverse events:
Cytokine release syndrome (CRS): A class effect of CD3-directed therapies. CRS occurs during the infusion course and is characterized by fever, nausea, headache, fatigue, myalgia, and other systemic symptoms. In teplizumab trials, CRS is generally mild to moderate and managed with premedication (acetaminophen and antihistamines) and supportive care during infusion. Severe CRS has been reported and requires prompt management. Infusion settings must be equipped to manage CRS.
Lymphopenia: T-cell depletion is expected with teplizumab and is part of its mechanism. Severe lymphopenia requires close monitoring. The most serious viral reactivation events observed in the clinical program occurred in patients who continued therapy despite persistent, severe lymphopenia. Current guidance requires dose interruption or discontinuation in the setting of persistent severe lymphopenia.
Viral reactivation: Serious, life-threatening cases of Epstein-Barr virus (EBV) and cytomegalovirus (CMV) reactivation have been reported. Most serious cases occurred in patients with persistent, severe lymphopenia who continued treatment. Viral reactivation surveillance before and during treatment is essential.
Other adverse events reported in PROTECT: Lymphopenia (expected), cytopenias (reductions in other blood cell types), gastrointestinal symptoms, rash, transaminase elevations, and headache. These were generally consistent with the expected immunomodulatory effects of the drug.
Pre-treatment requirements:
- Screening for EBV and CMV serostatus before initiating treatment
- Complete blood count monitoring before and during treatment courses
- Premedication before infusions to reduce CRS severity
- Avoid live vaccines during and after treatment until lymphocyte recovery is confirmed
Contraindications:
- Active serious infection
- Immunocompromised patients are at increased risk; careful benefit-risk assessment required
- Tzield is not effective as a disease-modifying therapy in non-autoimmune dysglycemic conditions: laboratory confirmation of autoimmune etiology (positive islet autoantibodies) is a clinical prerequisite
What This Means for Families and Clinicians
For families of a child recently diagnosed with T1D
If your child is aged 8 to 17 and has been diagnosed with type 1 diabetes within the past 8 weeks, Tzield is now an FDA-authorized option that deserves immediate discussion with your pediatric endocrinologist. This is not a therapy to consider “later.” The 8-week window is real and it closes.
Questions to ask your endocrinology team:
- Has my child had autoantibody testing that confirms autoimmune T1D?
- Are we within 8 weeks of diagnosis?
- Is Tzield appropriate for my child given their health history?
- Where can we receive the infusion courses?
- What is the prior authorization process with our insurance?
The Breakthrough T1D website and JDRF Tzield resources provide current information for families navigating this decision. Sanofi’s Tzield support program (1-800-633-1610) provides information about access, insurance navigation, and referral to treatment centers.
For pediatric endocrinologists
This approval creates a new, urgent clinical protocol: at the time of Stage 3 T1D diagnosis in any patient aged 8 to 17, the Tzield conversation should occur within the first visit or at minimum within the first week of diagnosis. This is the same urgency model used for conditions like acute lymphoblastic leukemia, where initiating disease-modifying treatment in the correct window fundamentally alters long-term outcomes.
The treatment infrastructure requirements (IV infusion over 12 consecutive days, monitoring for CRS and lymphopenia, EBV/CMV screening) mean that systems-level readiness at pediatric endocrinology centers is important. Centers not currently equipped to administer teplizumab should establish referral pathways to centers that are.
The broader significance
Dr. Aaron J. Kowalski, PhD, CEO of Breakthrough T1D, described the approval as providing “a novel therapy that targets the autoimmune and progressive nature of stage 3 type 1 diabetes,” noting that approximately 64,000 people are diagnosed with T1D every year in the United States. For the pediatric portion of that population, a brief window of opportunity now exists to alter the course of the disease in a way that was not previously possible.
Type 1 diabetes has been managed for over 100 years with increasingly sophisticated insulin replacement. This is the first time a treatment has been approved that goes beyond replacement to address the underlying autoimmune destruction. It is a genuinely historic moment for the T1D community.
For related HED coverage on other disease-modifying therapies and autoimmune conditions, see our post on Ocrevus (ocrelizumab) receiving pediatric approval for relapsing-remitting multiple sclerosis, another CD20-directed therapy that alters the course of an autoimmune disease, and our post on Awiqli, the first once-weekly basal insulin for type 2 diabetes, for context on how the insulin therapy landscape continues to evolve alongside disease-modifying approaches.
Sources
FDA approval announcement: FDA approves new indication for Tzield (teplizumab) for certain pediatric patients with recently diagnosed Stage 3 type 1 diabetes. FDA.gov. June 12 (announced June 15), 2026.
Sanofi press release: Sanofi’s Tzield approved in the US as the first disease-modifying therapy for patients recently diagnosed with stage 3 type 1 diabetes. Sanofi. June 12, 2026.
Drugs.com approval news: Sanofi’s Tzield Approved in the US as the First Disease-Modifying Therapy for Patients Recently Diagnosed with Stage 3 Type 1 Diabetes. drugs.com. June 13, 2026.
FDA plain language summary: FDA approves drug for pediatric stage 3 type I diabetes. FDA.gov. June 2026.
Pharmacy Times clinical review: FDA Approves Teplizumab-mzwv for Children With Newly Diagnosed Stage 3 Type 1 Diabetes. pharmacytimes.com. June 2026.
Patient Care Online clinical summary (with investigator cautions): FDA Expands Teplizumab Approval for Pediatric Stage 3 Type 1 Diabetes. patientcareonline.com. June 2026.
Healio endocrinology coverage: FDA approves Tzield to treat children and adolescents with stage 3 type 1 diabetes. healio.com. June 2026.
Pediatric Endocrine Society clinical summary: Teplizumab Approval Expanded to Stage 3 Type 1 Diabetes (Ages 8-17). pedsendo.org. June 2026.
Breakthrough T1D community update: Tzield approved for stage 3 T1D in the U.S. breakthrought1d.org. June 2026.
BioPharm International coverage: FDA Expands Pfizer’s Hympavzi Approval… (note: cross-reference; not directly cited for Tzield)
Sanofi Stage 2 expansion (April 2026, ages 1-7): Sanofi’s Tzield approved in the US to delay the onset of stage 3 type 1 diabetes in young children. sanofi.com. April 22, 2026.
PROTECT trial primary publication: Ramos EL, Dayan CM, Chatenoud L, et al. Teplizumab and beta-cell function in newly diagnosed type 1 diabetes. NEJM. 2023;389(23):2151–2161. doi:10.1056/NEJMoa2306691.
PROTECT trial registration: NCT03875729. ClinicalTrials.gov.
BETA-PRESERVE confirmatory trial registration: NCT05902884. ClinicalTrials.gov.
Teplizumab mechanism review: Teplizumab in Type 1 Diabetes. PMC9356435.
T1D staging scientific statement: Staging Presymptomatic Type 1 Diabetes. Diabetes. 2015;64(8):2541–2550.
T1D StatPearls: Type 1 Diabetes Mellitus. StatPearls. NCBI.
NIDDK T1D overview: Type 1 Diabetes. NIDDK.
Accelerated approval pathway: Accelerated Approval. FDA.gov.
EBV: Epstein-Barr Virus. StatPearls. NCBI.
CMV: Cytomegalovirus. StatPearls. NCBI.
Tzield prescribing information: TZIELD (teplizumab-mzwv) Prescribing Information. Sanofi. 2026.
Patient resources: Breakthrough T1D (JDRF) | American Diabetes Association: Type 1 | Sanofi Tzield patient support: 1-800-633-1610 | NIDDK Type 1 Diabetes
| Disclaimer: Health Evidence Digest provides general information about FDA approvals and health research for educational purposes. This content is not a substitute for professional medical advice. Tzield (teplizumab-mzwv) is indicated for recently diagnosed Stage 3 T1D in pediatric patients aged 8 to 17 years within 8 weeks of diagnosis. It is not effective in non-autoimmune dysglycemic conditions. Treatment requires IV infusion and monitoring in a qualified clinical setting. Families should consult with a board-certified pediatric endocrinologist immediately after a child’s Stage 3 T1D diagnosis to determine eligibility and timing within the treatment window. |
|---|